Піразольні сполуки як інгібітори sglt1
















Формула / Реферат
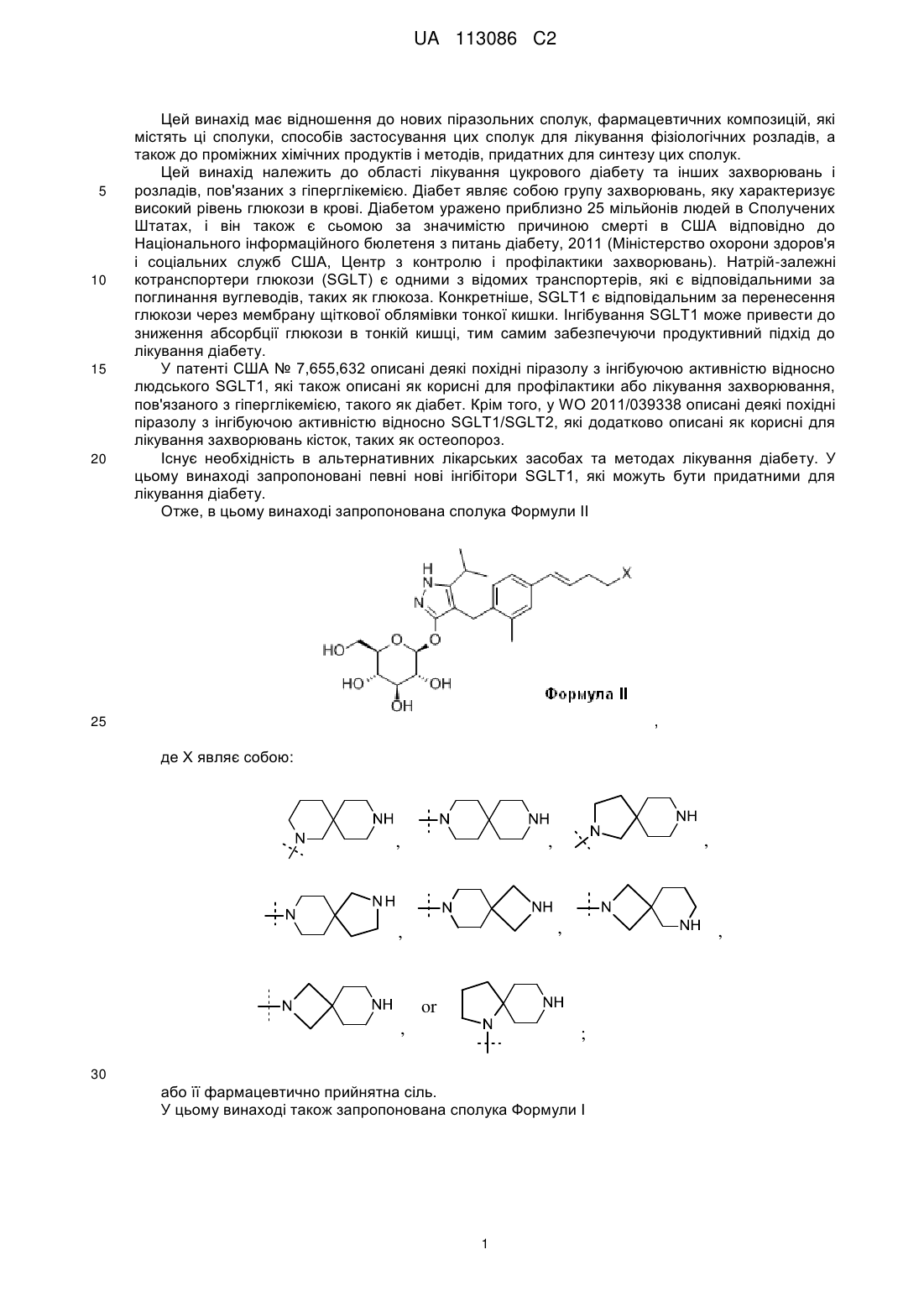
1. Сполука наведеної нижче формули:
 ,
,
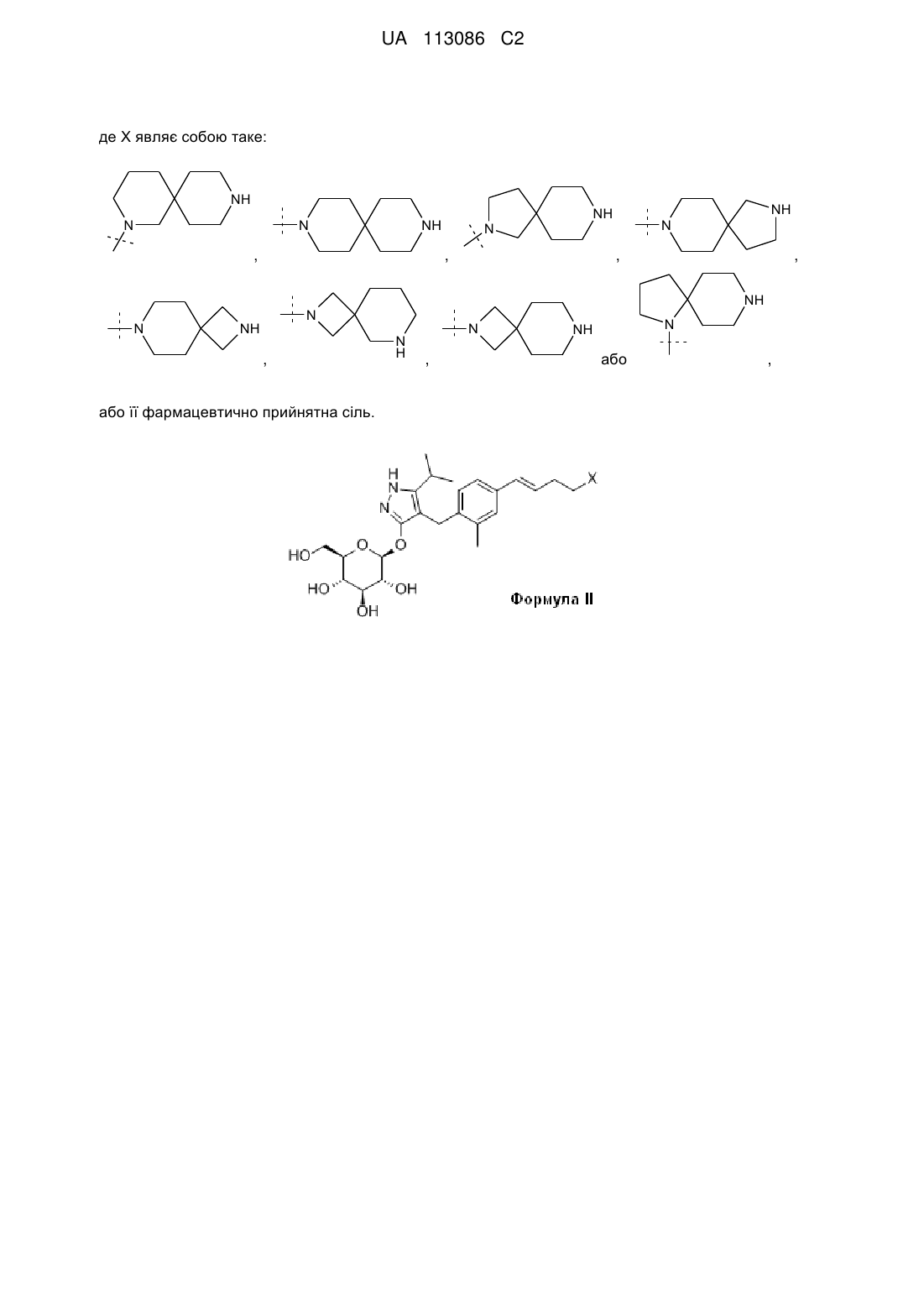
де X являє собою таке:
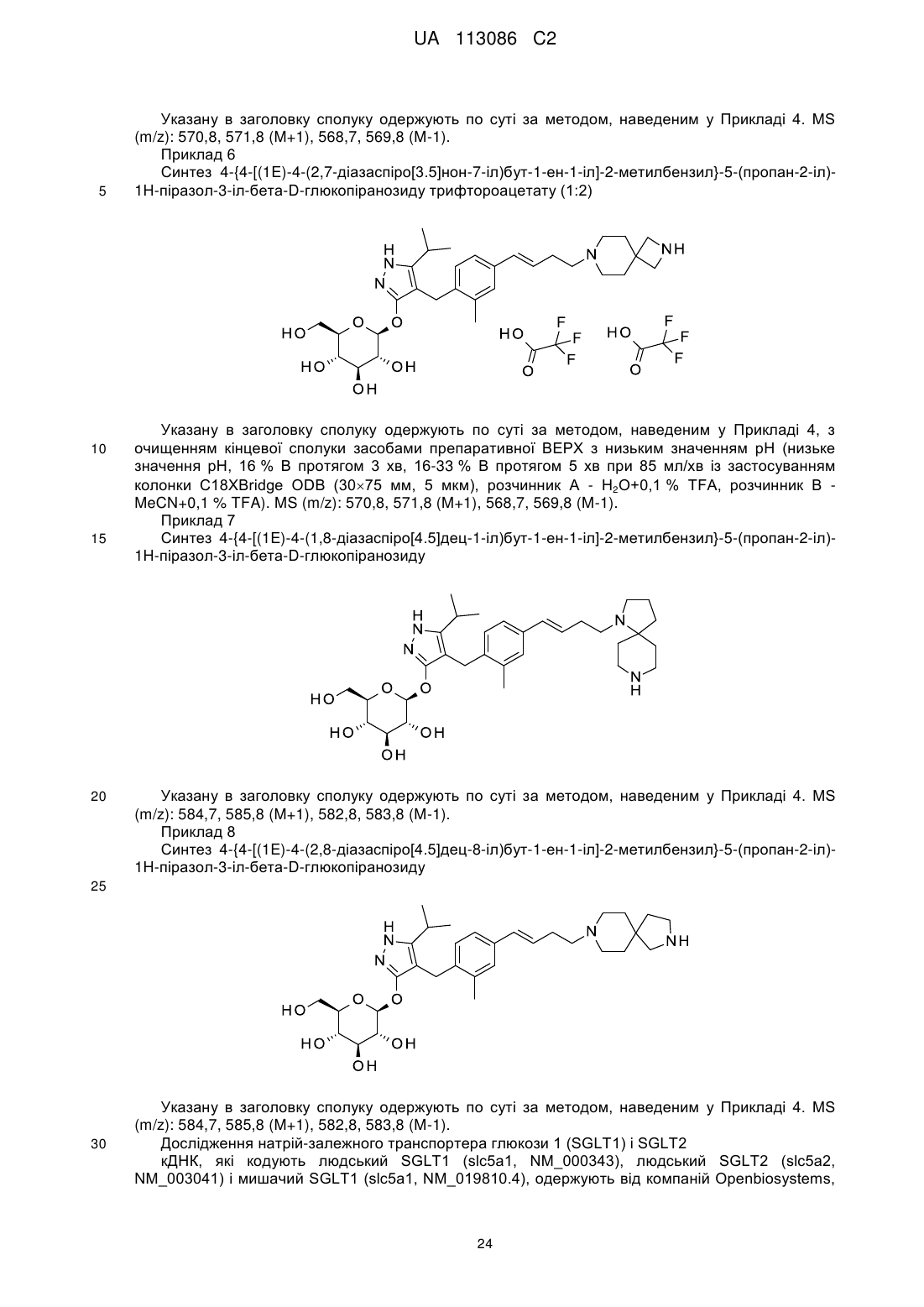
 ,
, 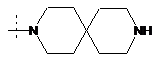 ,
, 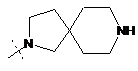 ,
, 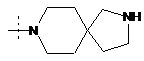 ,
, 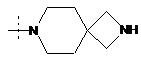 ,
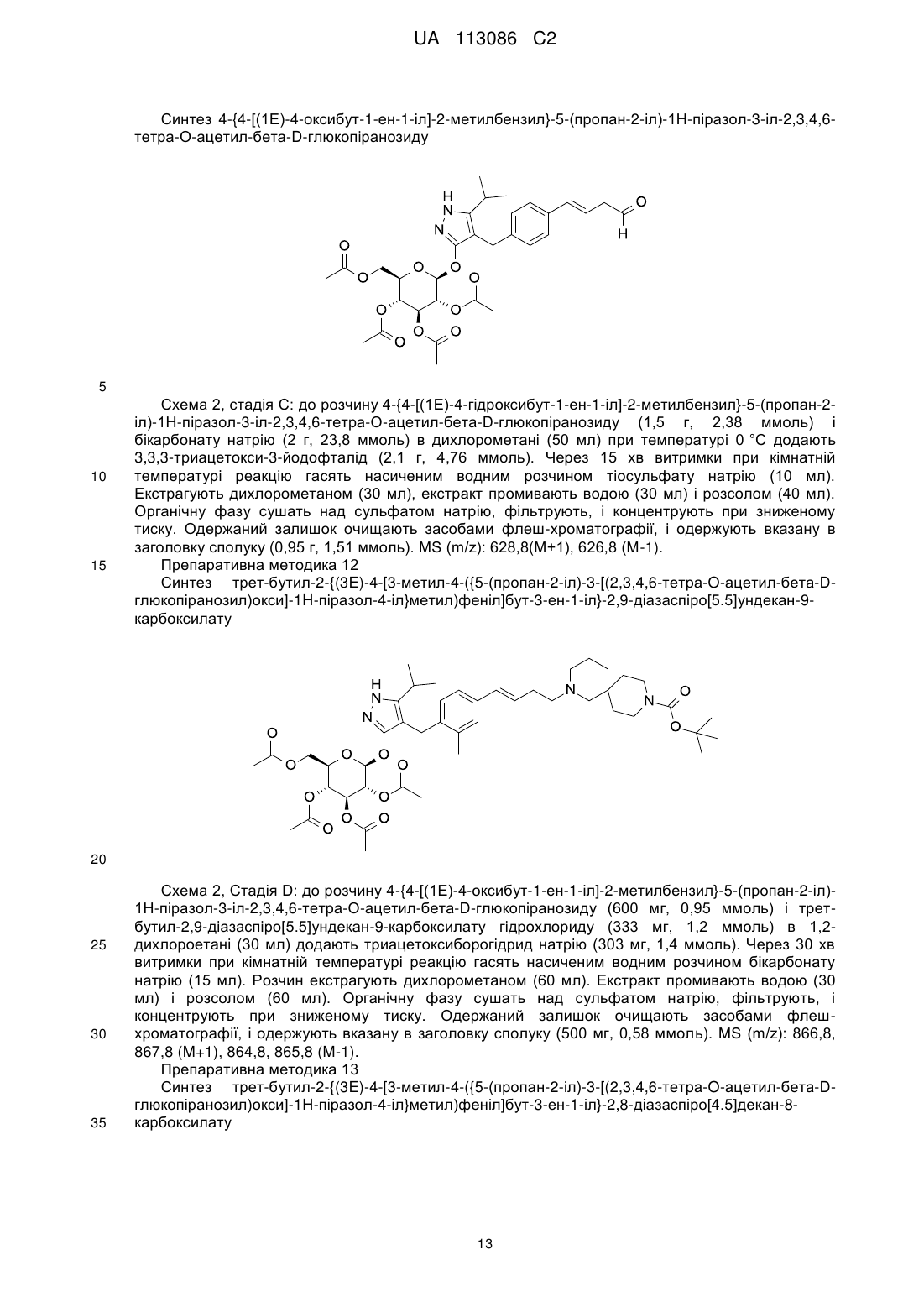
, 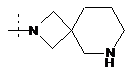 ,
, 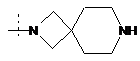 або
або 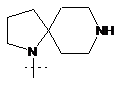 ,
,
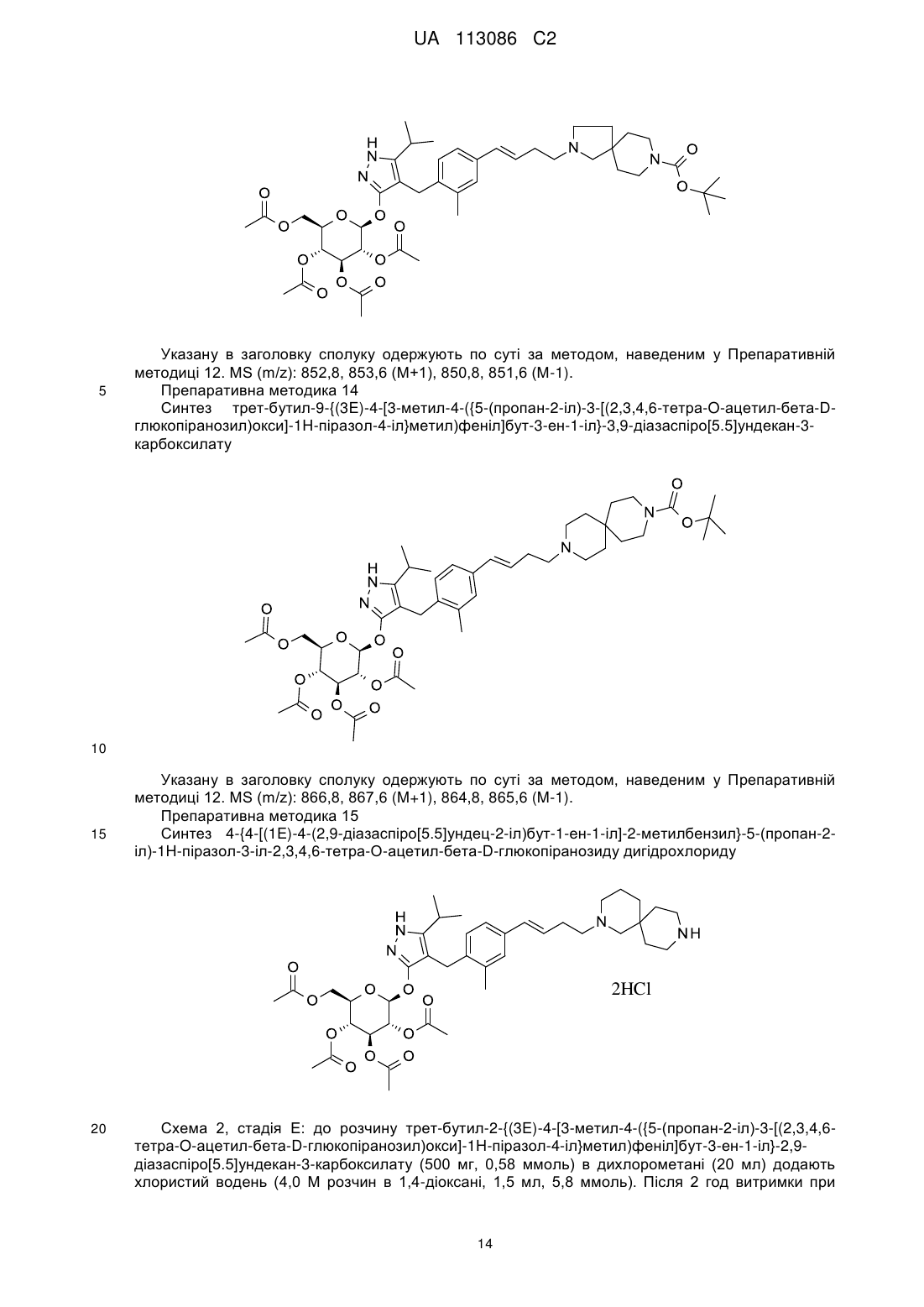
або її фармацевтично прийнятна сіль.
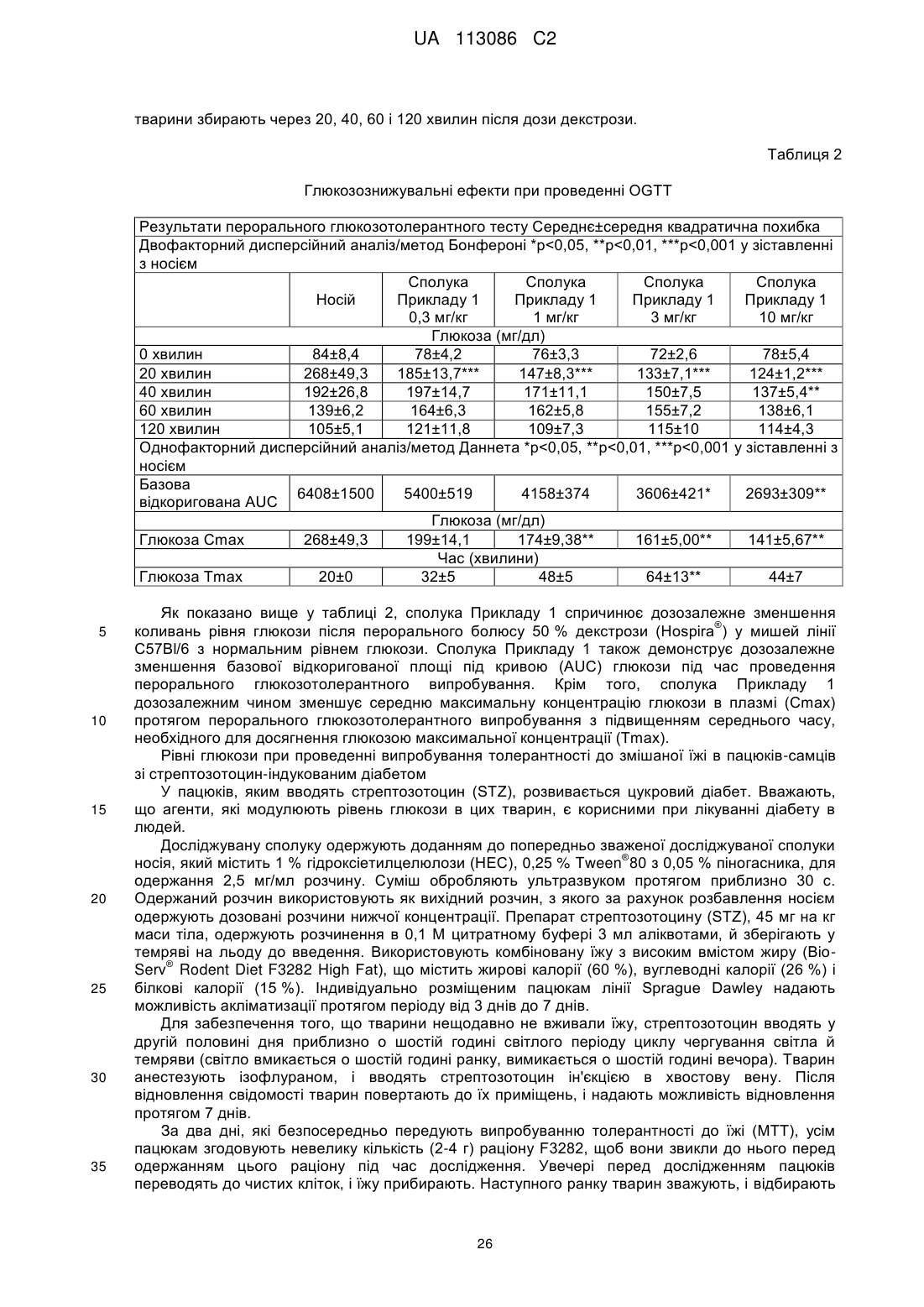
2. Сполука або сіль за п. 1, яка являє собою:
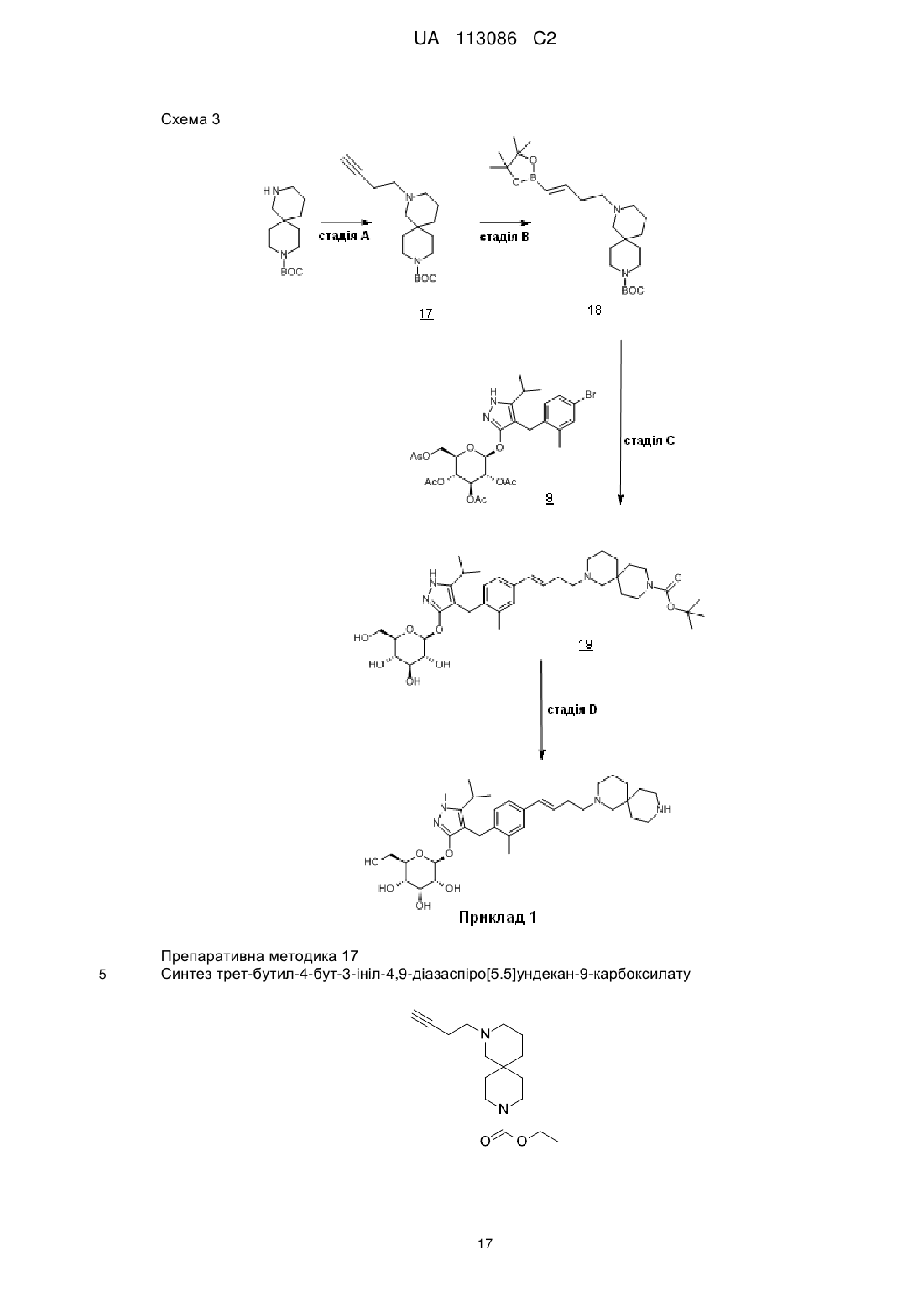
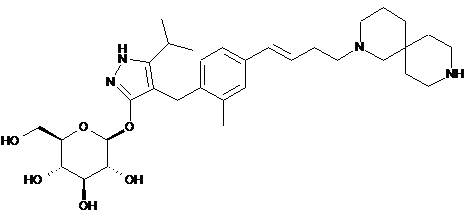 .
.
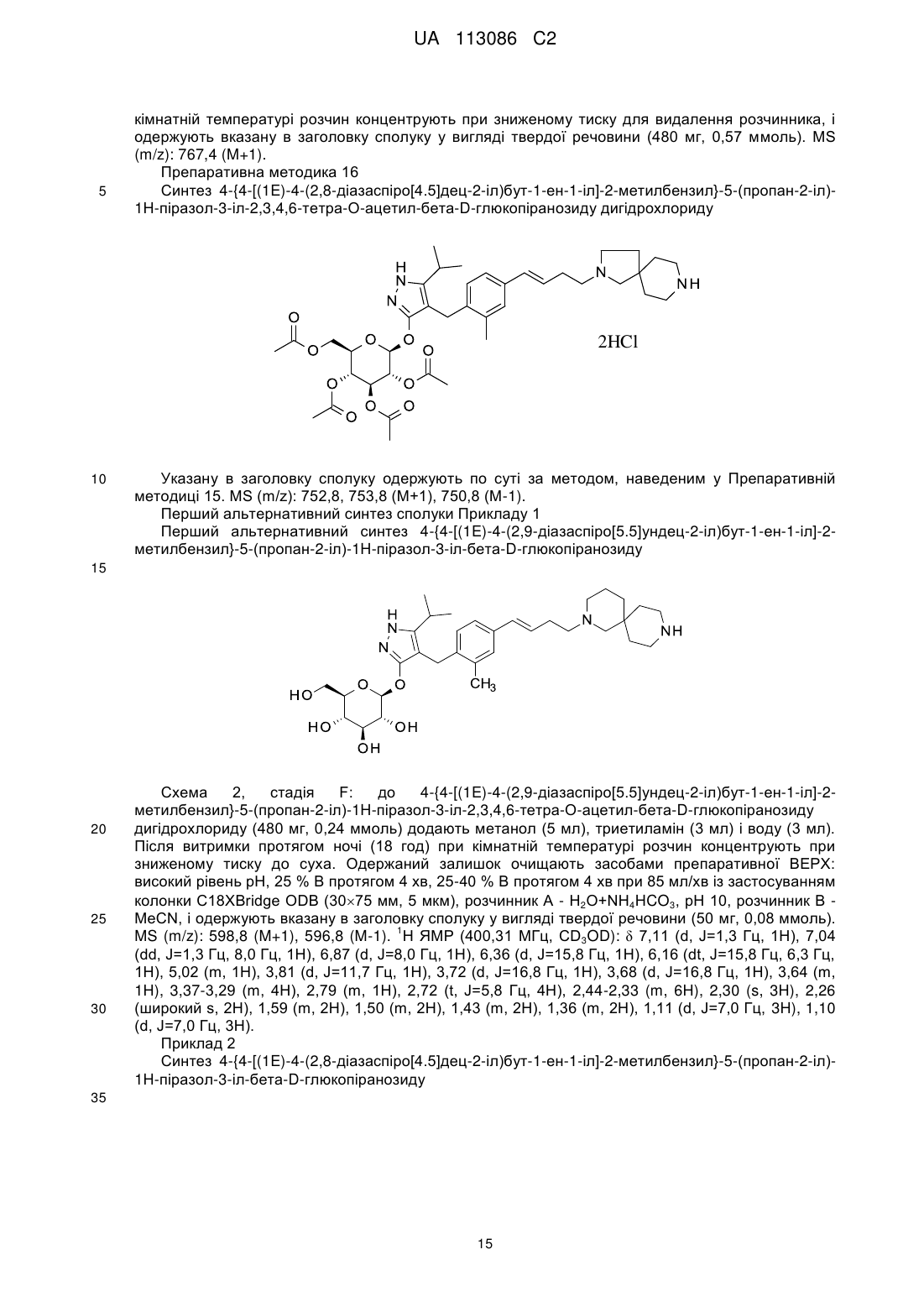
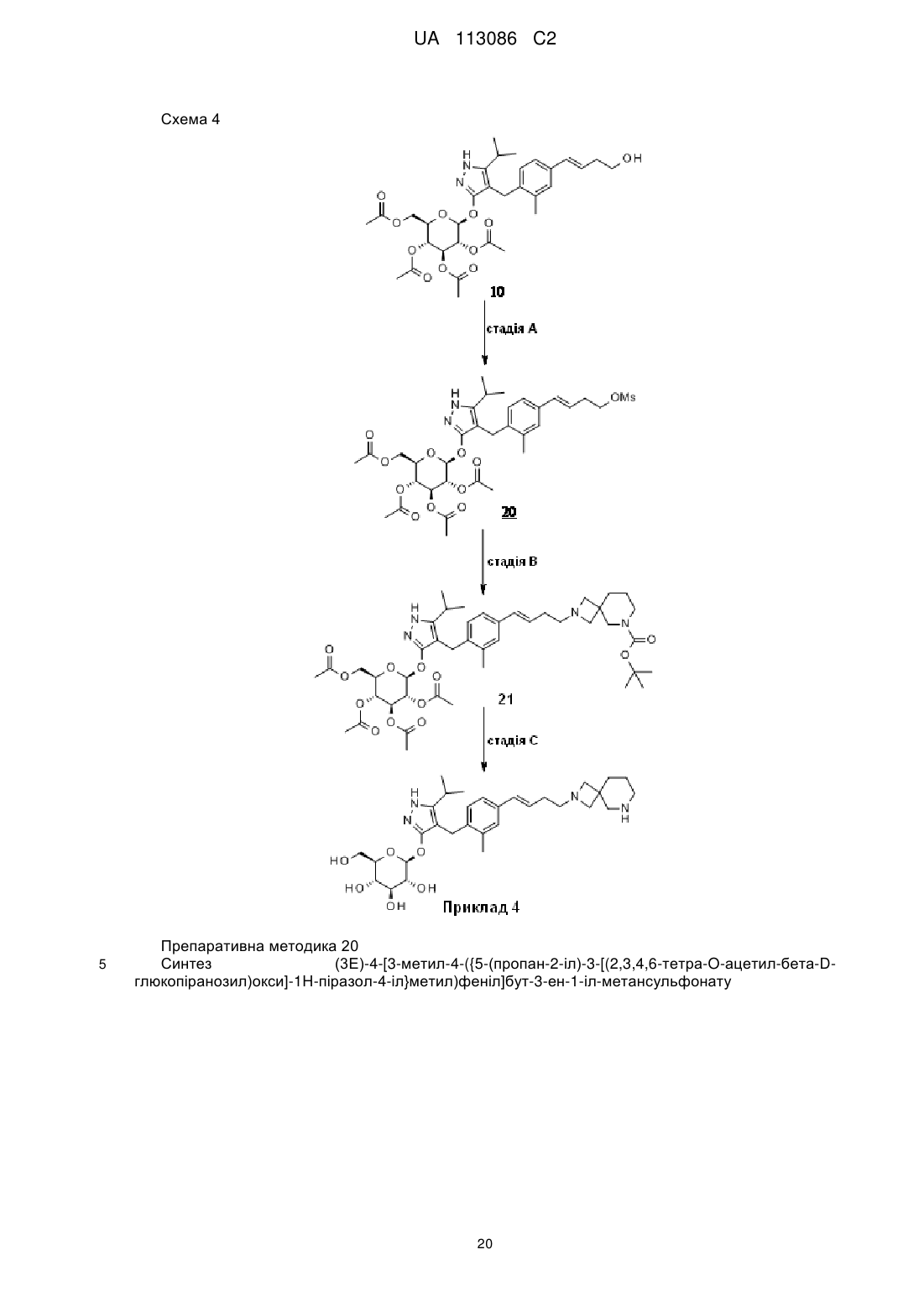
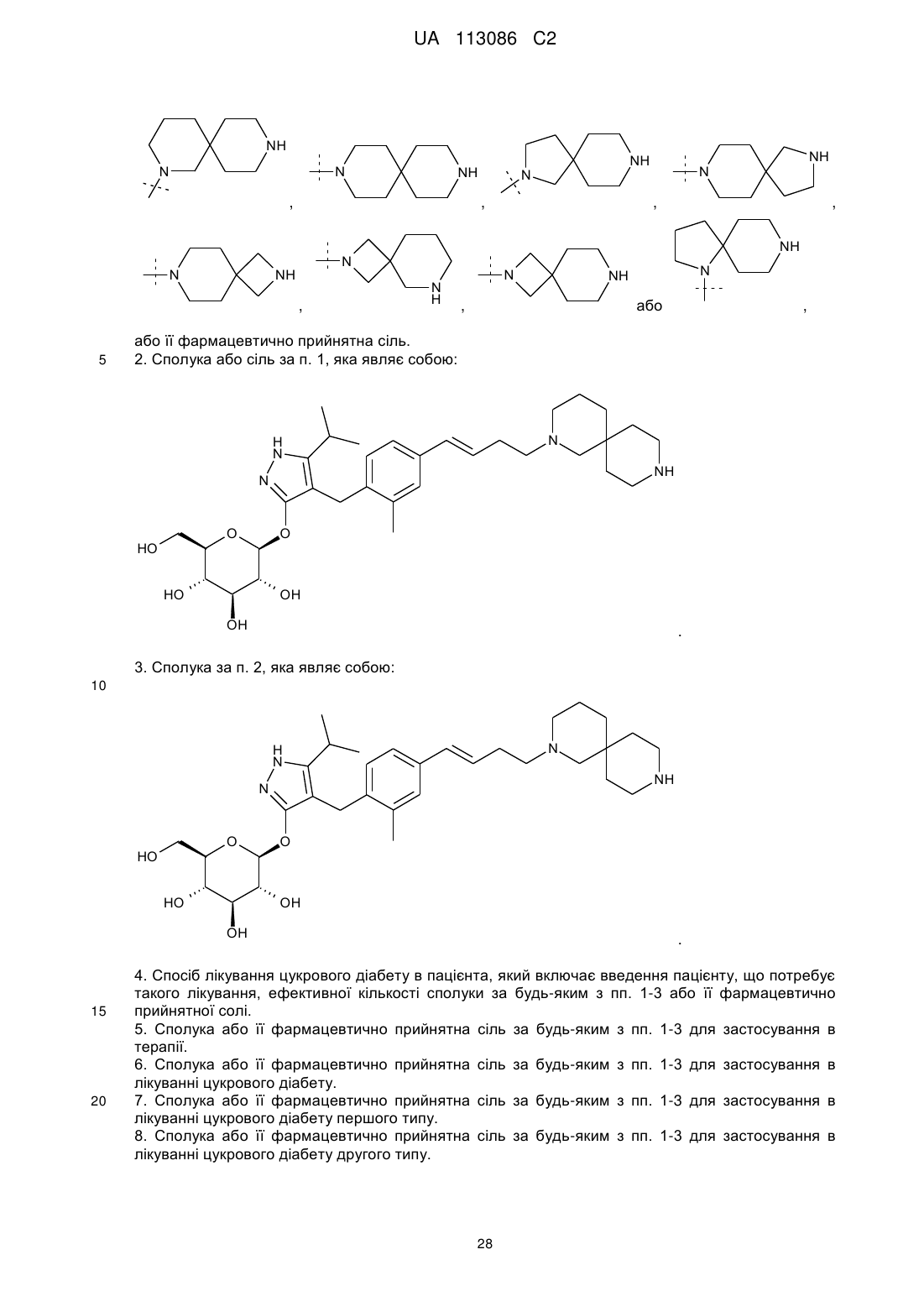
3. Сполука за п. 2, яка являє собою:
.
4. Спосіб лікування цукрового діабету в пацієнта, який включає введення пацієнту, що потребує такого лікування, ефективної кількості сполуки за будь-яким з пп. 1-3 або її фармацевтично прийнятної солі.
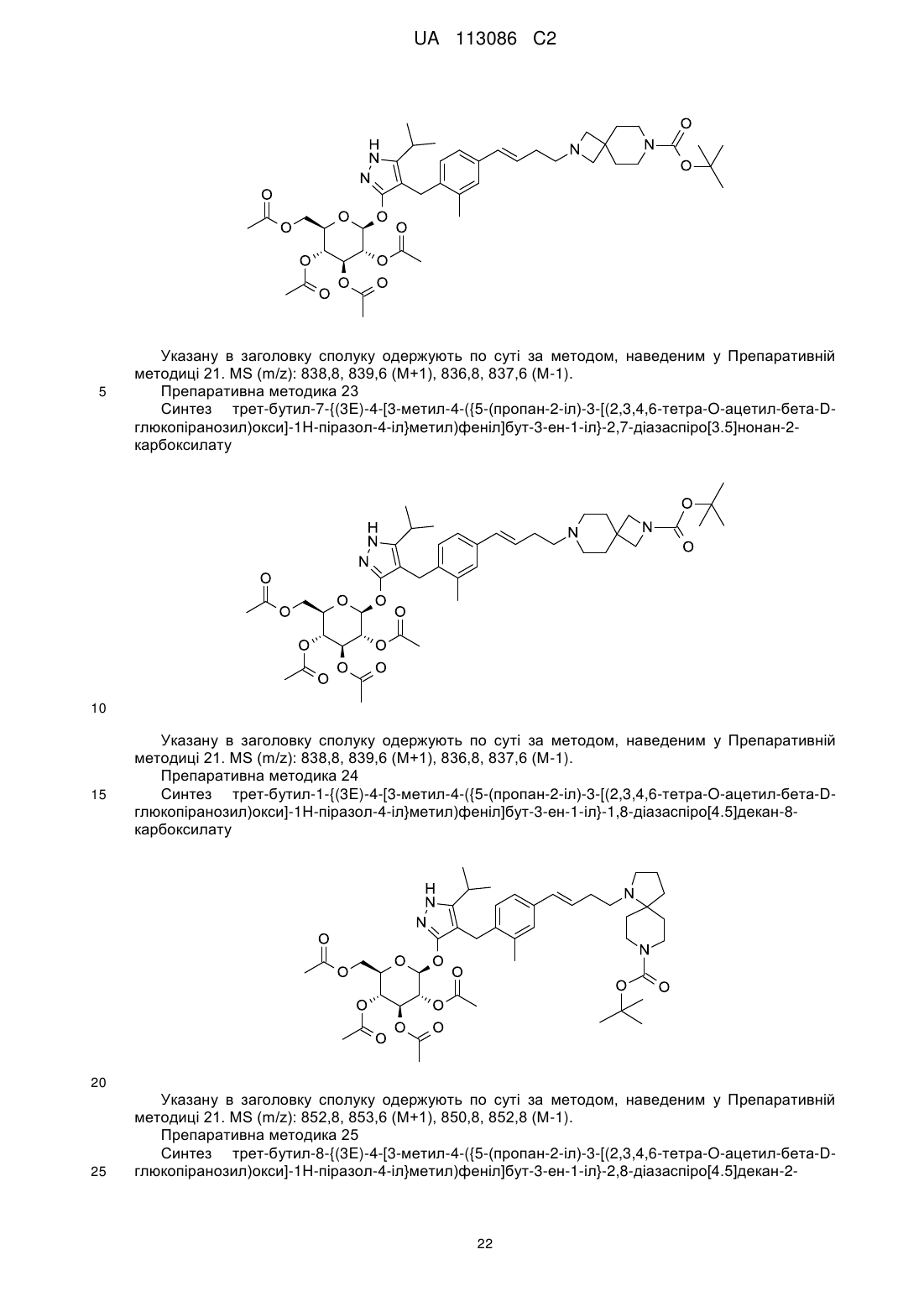
5. Сполука або її фармацевтично прийнятна сіль за будь-яким з пп. 1-3 для застосування в терапії.
6. Сполука або її фармацевтично прийнятна сіль за будь-яким з пп. 1-3 для застосування в лікуванні цукрового діабету.
7. Сполука або її фармацевтично прийнятна сіль за будь-яким з пп. 1-3 для застосування в лікуванні цукрового діабету першого типу.
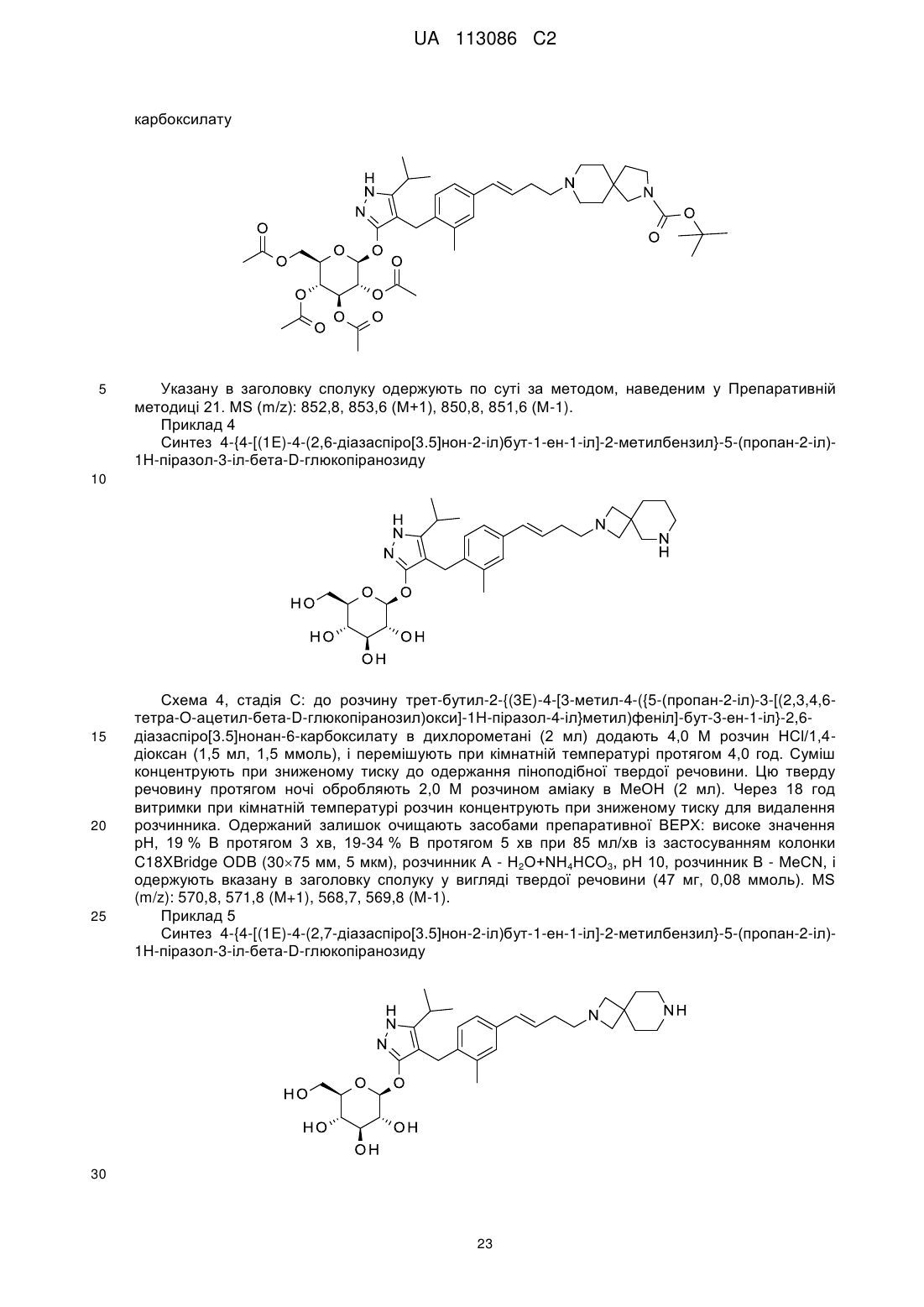
8. Сполука або її фармацевтично прийнятна сіль за будь-яким з пп. 1-3 для застосування в лікуванні цукрового діабету другого типу.
9. Застосування сполуки або її фармацевтично прийнятної солі за будь-яким з пп. 1-3 для виготовлення лікарського засобу для лікування цукрового діабету.
10. Застосування сполуки або її фармацевтично прийнятної солі за будь-яким з пп. 1-3 для виготовлення лікарського засобу для лікування цукрового діабету першого типу.
11. Застосування сполуки або її фармацевтично прийнятної солі за будь-яким з пп. 1-3 для виготовлення лікарського засобу для лікування цукрового діабету другого типу.
12. Фармацевтична композиція, яка містить сполуку або її фармацевтично прийнятну сіль за будь-яким з пп. 1-3 в комбінації з одним або більше фармацевтично прийнятними носіями, розріджувачами або наповнювачами.
13. Фармацевтична композиція за п. 12, яка містить один або більше інших терапевтичних засобів.
Текст
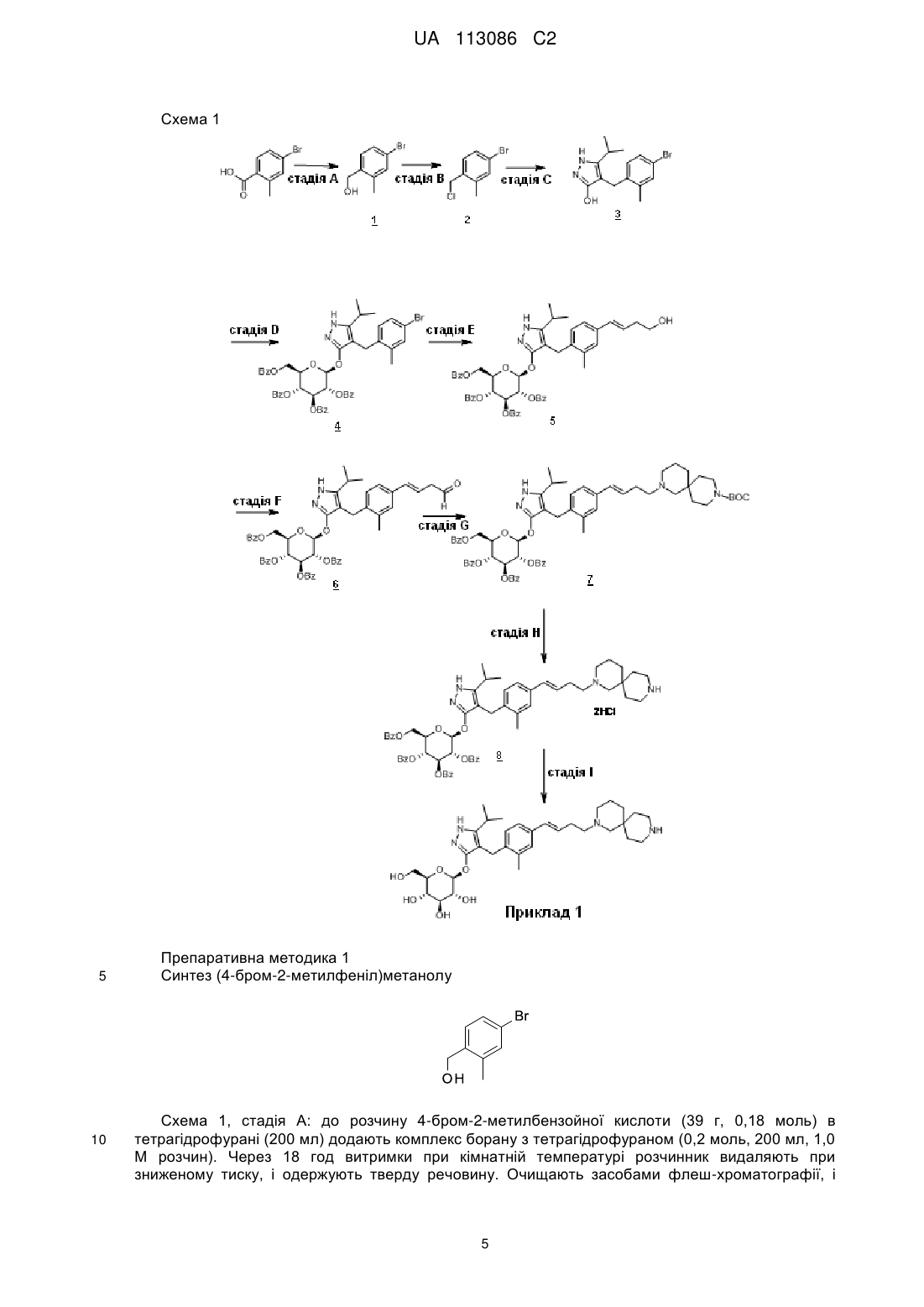
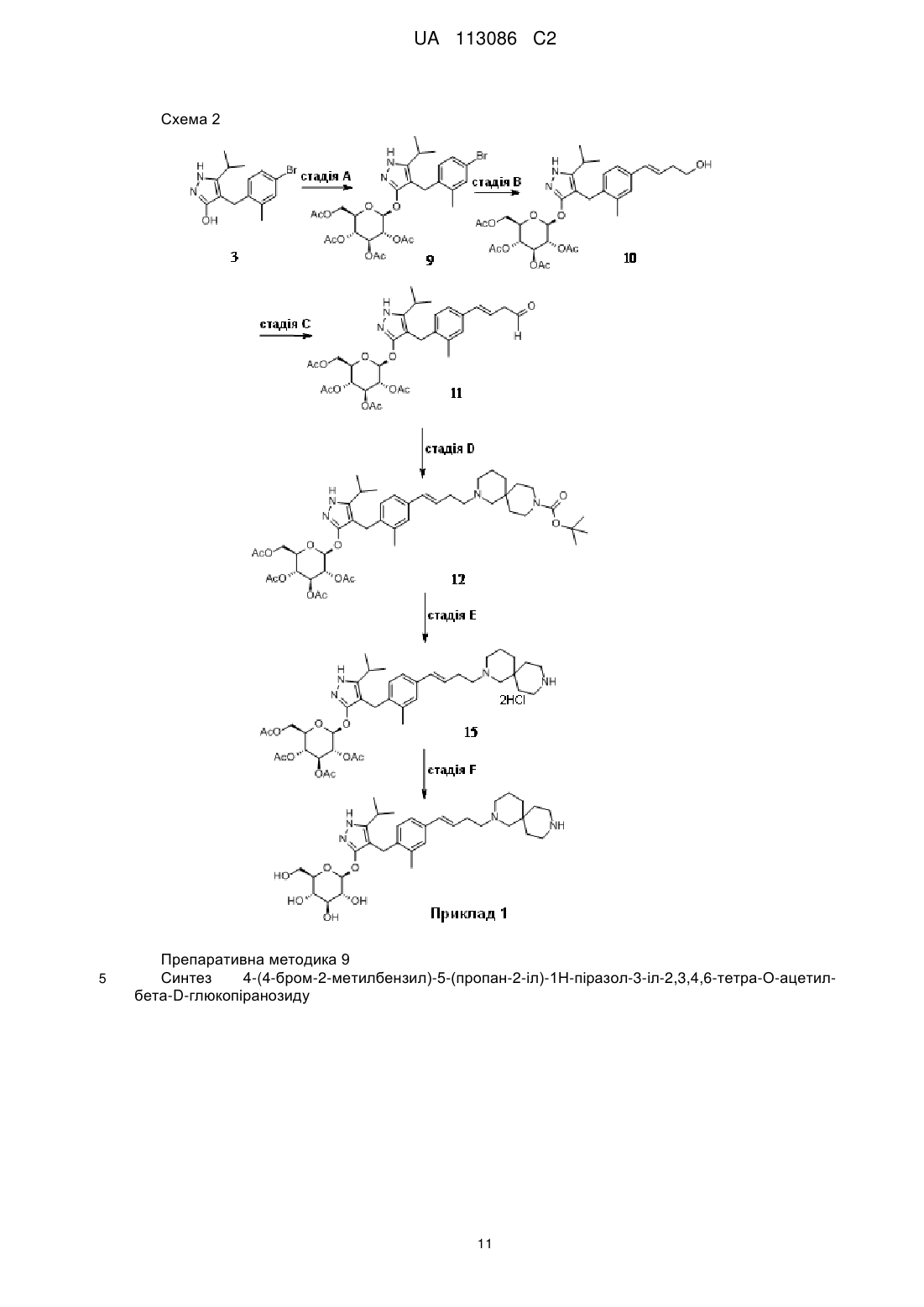
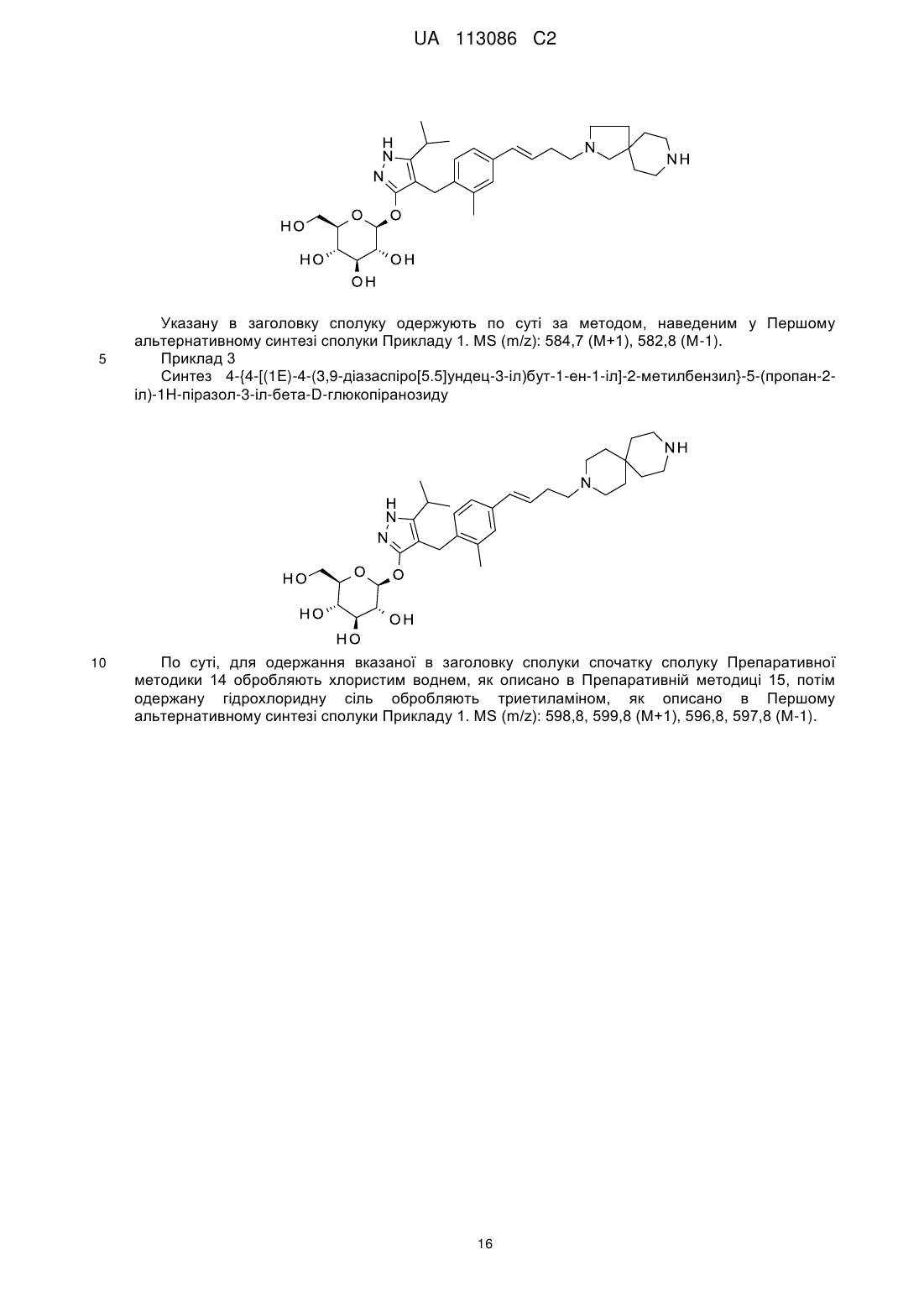
Реферат: У цьому винаході запропонована сполука Формули II: H N X N O HO HO O OH OH , UA 113086 C2 (12) UA 113086 C2 де X являє собою таке: NH N N NH , N N , , або її фармацевтично прийнятна сіль. N , , NH N NH NH NH N H N , N NH або , UA 113086 C2 5 10 15 20 Цей винахід має відношення до нових піразольних сполук, фармацевтичних композицій, які містять ці сполуки, способів застосування цих сполук для лікування фізіологічних розладів, а також до проміжних хімічних продуктів і методів, придатних для синтезу цих сполук. Цей винахід належить до області лікування цукрового діабету та інших захворювань і розладів, пов'язаних з гіперглікемією. Діабет являє собою групу захворювань, яку характеризує високий рівень глюкози в крові. Діабетом уражено приблизно 25 мільйонів людей в Сполучених Штатах, і він також є сьомою за значимістю причиною смерті в США відповідно до Національного інформаційного бюлетеня з питань діабету, 2011 (Міністерство охорони здоров'я і соціальних служб США, Центр з контролю і профілактики захворювань). Натрій-залежні котранспортери глюкози (SGLT) є одними з відомих транспортерів, які є відповідальними за поглинання вуглеводів, таких як глюкоза. Конкретніше, SGLT1 є відповідальним за перенесення глюкози через мембрану щіткової облямівки тонкої кишки. Інгібування SGLT1 може привести до зниження абсорбції глюкози в тонкій кишці, тим самим забезпечуючи продуктивний підхід до лікування діабету. У патенті США № 7,655,632 описані деякі похідні піразолу з інгібуючою активністю відносно людського SGLT1, які також описані як корисні для профілактики або лікування захворювання, пов'язаного з гіперглікемією, такого як діабет. Крім того, у WO 2011/039338 описані деякі похідні піразолу з інгібуючою активністю відносно SGLT1/SGLT2, які додатково описані як корисні для лікування захворювань кісток, таких як остеопороз. Існує необхідність в альтернативних лікарських засобах та методах лікування діабету. У цьому винаході запропоновані певні нові інгібітори SGLT1, які можуть бути придатними для лікування діабету. Отже, в цьому винаході запропонована сполука Формули II , 25 де X являє собою: , , , , , , or , ; 30 або її фармацевтично прийнятна сіль. У цьому винаході також запропонована сполука Формули I 1 UA 113086 C2 5 10 15 20 25 30 35 40 45 50 або її фармацевтично прийнятна сіль. У цьому винаході також запропонований спосіб лікування цукрового діабету в пацієнта, і цей спосіб включає введення пацієнту, який потребує такого лікування, ефективної кількості сполуки Формули I або сполуки Формули II чи фармацевтично прийнятної солі цих сполук. У цьому винаході також запропонований спосіб лікування цукрового діабету першого типу в пацієнта, що включає введення пацієнту, який потребує такого лікування, ефективної кількості сполуки Формули I або Формули II чи фармацевтично прийнятної солі цієї сполуки. Крім того, у цьому винаході запропонований спосіб лікування цукрового діабету другого типу в пацієнта, що включає введення пацієнту, який потребує такого лікування, ефективної кількості сполуки Формули I або Формули II чи фармацевтично прийнятної солі такої сполуки. У цьому винаході також запропонований спосіб лікування порушення толерантності до глюкози (IGT), порушення рівня глюкози крові натщесерце (IFG) або метаболічного синдрому в пацієнта, і цей спосіб включає введення пацієнту, який потребує такого лікування, ефективної кількості сполуки Формули I або Формули II чи фармацевтично прийнятної солі такої сполуки. Крім того, у цьому винаході запропонована сполука Формули I або Формули II чи фармацевтично прийнятна сіль цієї сполуки для застосування в терапії, зокрема для лікування цукрового діабету. Крім того, у цьому винаході запропонована сполука Формули I або Формули II чи фармацевтично прийнятна сіль цієї сполуки для застосування в лікуванні цукрового діабету першого типу. Крім того, у цьому винаході запропонована сполука Формули I або Формули II чи фармацевтично прийнятна сіль цієї сполуки для застосування в лікуванні цукрового діабету другого типу. У цьому винаході також запропоноване застосування сполуки Формули I або Формули II чи фармацевтично прийнятної солі цієї сполуки для виготовлення лікарського засобу для лікування цукрового діабету. Крім того, у цьому винаході запропоноване застосування сполуки Формули I або Формули II чи фармацевтично прийнятної солі цієї сполуки для виготовлення лікарського засобу для лікування цукрового діабету першого типу. У цьому винаході також запропоноване застосування сполуки Формули I або Формули II чи фармацевтично прийнятної солі цієї сполуки для виготовлення лікарського засобу для лікування цукрового діабету другого типу. У цьому винаході також запропоноване застосування сполуки Формули I або Формули II чи фармацевтично прийнятної солі цієї сполуки для виготовлення лікарського засобу для лікування порушення толерантності до глюкози (IGT), порушення рівня глюкози крові натщесерце (IFG) або метаболічного синдрому. У цьому винаході також запропонована фармацевтична композиція, яка містить сполуку Формули I або Формули II, чи фармацевтично прийнятну сіль цієї сполуки, в комбінації з одним або більше фармацевтично прийнятним(-ми) носієм(-ями), розріджувачем(-ами) або наповнювачем(-ами). У певному варіанті здійснення ця композиція містить також один або більше інший(-их) терапевтичний(-их) засіб(-ів). У цьому винаході також охоплені нові проміжні хімічні продукти й способи синтезу сполуки Формули I або Формули II. Термін "лікування" (або "лікувати"), вжитий в цьому описі, означає перешкоджання, стримування, уповільнення, зупинку або звертання розвитку або тяжкості існуючого симптому або розладу. Термін "пацієнт", вжитий в цьому описі, означає ссавця, наприклад, мишу, морську свинку, пацюка, собаку або людину. Зрозуміло, що пацієнтом переважно є людина. Термін "ефективна кількість", вжитий в цьому описі, означає таку кількість або дозу сполуки за цим винаходом чи фармацевтично прийнятної солі цієї сполуки, яка після одноразового або багаторазового введення пацієнту забезпечує бажаний ефект у пацієнта при призначенні лікування або лікуванні. Ефективна кількість може бути легко визначена лікарем, який призначає лікування, як фахівцем у цій галузі, із застосуванням відомих методик і внаслідок спостереження результатів, 2 UA 113086 C2 5 10 15 20 25 30 35 40 45 одержаних за аналогічних обставин. При визначенні ефективної для пацієнта кількості лікар, який призначає лікування, розглядає низку факторів, у тому числі, але не обмежуючись ними: вид ссавця; розмір, вік і загальний стан здоров'я особини; конкретне захворювання або розлад, яким уражений пацієнт; ступінь чи ураження, чи тяжкість захворювання або розладу; індивідуальна реакція пацієнта; конкретна призначена сполука; спосіб введення; характеристики біодоступності введеного препарату; вибрана схема прийому лікарського засобу; використання супутнього лікарського засобу; та інші важливі обставини. Сполуки Формули I і Формули II зазвичай є ефективними в широкому діапазоні доз. Наприклад, добові дози зазвичай становлять від приблизно 0,01 мг на кг маси тіла до приблизно 30 мг на кг маси тіла. У деяких випадках дози нижче нижньої межі згаданого діапазону можуть бути більш ніж достатніми, тоді як в інших випадках можуть бути використані ще більш високі дози без спричинення жодних шкідливих побічних ефектів, і, отже, згаданий діапазон доз жодним чином не є призначеним для обмеження обсягу цього винаходу. Сполуки за цим винаходом за варіантом, якому віддають перевагу, виготовляють у вигляді фармацевтичних композицій, які вводять будь-яким способом, який забезпечує біологічну активність цієї сполуки. За варіантом, якому віддають найбільшу перевагу, такі композиції призначені для перорального введення. Такі фармацевтичні композиції і способи їх одержання є добре відомими в цій галузі (дивись, наприклад, Remington: The Science and Practice of Pharmacy (Troy, D.B. Editor, 21st Edition, Lippincott, Williams & Wilkins, 2006)). За ще одним із аспектів цього винаходу згадані сполуки вводять у комбінації з одним або більше терапевтичним(-ми) засобом(-ами), таким(-ми) як протидіабетичні засоби. Введення в комбінації включає одночасне або послідовне введення. Крім того, одночасне введення згаданої комбінації може відбуватися у вигляді разової комбінованої дози або окремих доз кожного терапевтичного засобу. До прикладів протидіабетичних засобів належать метформін (metformin); інгібітор DPPIV (дипептидилпептидаза IV), такий як сітагліптин (sitagliptin) або лінагліптин (linagliptin); сульфонілсечовина, така як глімепірид (glimepiride); тіазолідиндіон, такий як піоглітазон (pioglitazone); базальний інсулін, такий як гларгін (glargine); швидкодіючий інсулін, такий як HUMALOG або NOVOLOG; агоніст GLP-1 (глюкагоноподібний пептид), такий як екзенатид (exеnatide) або ліраглутид (liraglutide); інгібітор SGLT2, такий як дапагліфлозин (dapagliflozin) або емпагліфлозин (empagliflozin); антагоніст рецептора глюкагону, такий як LY2409021 тощо. Сполуки Формули I і Формули II одержують, як показано в прикладах і на схемах, наведених нижче. Реагенти та вихідні матеріали є легко доступними фахівцеві в цій галузі. Усі замісники, якщо не зазначено інше, єтакими, як визначено вище. Зрозуміло, що наведені схеми, препаративні методики та приклади жодним чином не призначені для обмеження обсягу цього винаходу. До прикладів способів розділення належать селективні методи кристалізації або хіральна хроматографія (дивись, наприклад, Jacques, J., et al. "Еnantiomers, Racemates, and Resolutions", John Wiley and Sons, Inc., 1981 та Eliel, E.L. and Wilеn, S.H.,"Stereochemistry of Organic Compounds", Wiley-Intersciеnce, 1994). Фахівець в цій галузі має розуміти, що розділення та виділення окремих діастереомерів або геометричних ізомерів сполук Формули I чи Формули II, або окремих діастереомерів чи геометричних ізомерів проміжних хімічних сполук, що ведуть до одержання сполук Формули I або Формули II, засобами хроматографії, хіральної хроматографії або селективної кристалізації може відбуватися в будь-якій зручній точці процесу синтезу. Позначка "δ", вжита в цьому описі, означає частину на мільйон у бік слабкого поля від тетраметилсилану; "хв" означає хвилину або хвилини; "THF" означає тетрагідрофуран; "МеОН" означає метанол або метиловий спирт; "ВЕРХ" означає високоефективну рідинну хроматографію; "Ас" означає ацетиловий замісник наведеної нижче структури: . 50 Позначка "Bz" означає бензоїльний замісник наведеної нижче структури: 3 UA 113086 C2 . 5 10 Термін "ВОС" означає трет-бутилоксикарбонільну захисну групу. Фармацевтично прийнятні солі й загальна методика їх одержання є добре відомими в цій галузі (дивись, наприклад, Gould, P.L., "Salt selection for basic drugs", International Journal of Pharmaceutics, 33: 201-217 (1986); Bastin et al. "Salt Selection and Optimization Procedures for Pharmaceutical New Chemical Еntities", Organic Process Research and Developmеnt, 4: 427-435 (2000) та Berge, S.M., et al., "Pharmaceutical Salts", Journal of Pharmaceutical Sciеnces, Vol. 66, No. 1, January 1977). Фахівцеві в галузі синтезу буде зрозуміло, що сполуки Формули I і Формули II, такі як аміни, є органічними основами, і що вони можуть бути легко перетворені на фармацевтично прийнятні солі та виділені у вигляді фармацевтично прийнятних солей, таких як тартратна сіль або хлористоводнева сіль, із застосуванням методів й умов, добре відомих фахівцеві в цій галузі. 4 UA 113086 C2 Схема 1 5 10 Препаративна методика 1 Синтез (4-бром-2-метилфеніл)метанолу Схема 1, стадія А: до розчину 4-бром-2-метилбензойної кислоти (39 г, 0,18 моль) в тетрагідрофурані (200 мл) додають комплекс борану з тетрагідрофураном (0,2 моль, 200 мл, 1,0 М розчин). Через 18 год витримки при кімнатній температурі розчинник видаляють при зниженому тиску, і одержують тверду речовину. Очищають засобами флеш-хроматографії, і 5 UA 113086 C2 5 10 15 20 25 30 35 40 одержують вказану в заголовку сполуку у вигляді твердої речовини білого кольору (32,9 г, 0,16 1 моль). H ЯМР (CDCl3): δ 1,55 (s, 1H), 2,28 (s, 3H), 4,61 (s, 2H), 7,18-7,29 (m, 3H). Альтернативний синтез (4-бром-2-метилфеніл)метанолу До розчину 4-бром-2-метилбензойної кислоти (24,3 г, 0,113 моль) у безводному тетрагідрофурані (THF, 146 мл) при температурі 3 °C повільно додають комплекс борану з диметилсульфідом (2 М розчин в тетрагідрофурані, 116 мл, 0,232 моль). Після перемішування на холоді протягом 10 хв охолоджувальну баню видаляють, і реакційну суміш витримують для повільного нагрівання до кімнатної температури. Через 1 год розчин охолоджують до температури 5 °C, і повільно додають воду (100 мл). Додають етилацетат (100 мл), і фази відокремлюють. Органічний шар промивають насиченим водним розчином NaHCO 3 (200 мл), і сушать над Na2SO4. Після фільтрування й концентрування при зниженому тиску одержують залишок, який очищають фільтруванням через тонкий шар діоксиду кремнію, елююють 15 % розчином етилацетат/ізогексан, і одержують вказану в заголовку сполуку (20,7 г, вихід 91,2 %). MS (m/z): 183/185 (M+1-18). Препаративна методика 2 Синтез 4-бром-1-хлорометил-2-метилбензолу Схема 1, стадія В: до розчину (4-бром-2-метилфеніл)метанолу (32,9 г, 0,16 моль) в дихлорометані (200 мл) і диметилформаміді (0,025 моль, 2,0 мл) при температурі 0 °C додають тіонілхлорид (14,31 мл, 0,2 моль). Через 1 год витримки при кімнатній температурі цю суміш вливають в льодоводяну суміш (100 г), екстрагують дихлорометаном (300 мл), екстракт промивають 5 % водним розчином бікарбонату натрію (30 мл) і сольовим розчином (200 мл), сушать над сульфатом натрію, концентрують при зниженому тиску, і одержують неочищену вказану в заголовку сполуку у вигляді твердої речовини білого кольору (35,0 г, 0,16 моль). Цей 1 матеріал використовують на наступній стадії реакції без додаткового очищення. H ЯМР (CDCl3): δ 2,38 (s, 3H), 4,52 (s, 2H), 7,13-7,35 (m, 3H). Альтернативний синтез 4-бром-1-хлорметил-2-метилбензолу До охолодженого в льодоводяній суміші розчину (4-бром-2-метилфеніл)метанолу (16,14 г, 80,27 ммоль) і триетиламіну (16,78 мл, 120,4 ммоль) в дихлорометані (80,7 мл) повільно додають метансульфонілхлорид (6,83 мл, 88,3 ммоль). Цю суміш лишають для повільного нагрівання до кімнатної температури, і перемішують протягом 16 год. Додають ще метансульфонілхлорид (1,24 мл, 16,1 ммоль), і суміш перемішують при кімнатній температурі протягом 2 год. Додають воду (80 мл), і фази відокремлюють. Органічний шар промивають соляною кислотою (1 н. розчин; 80 мл), потім насиченим водним розчином гідрокарбонату натрію (80 мл), потім водою (80 мл), і сушать над Na 2SO4. Після фільтрування й концентрування при зниженому тиску одержують залишок, який очищають засобами флеш-хроматографії (з 1 елююванням гексаном), і одержують вказану в заголовку сполуку (14,2 г, вихід 80,5 %). H ЯМР (300,11 МГц, CDCl3): δ 7,36-7,30 (m, 2H), 7,18 (d, J=8,1 Гц, 1H), 4,55 (s, 2H), 2,41 (s, 3H). Препаративна методика 3 Синтез 4-[(4-бром-2-метилфеніл)метил]-5-ізопропіл-1Н-піразол-3-олу 45 50 Схема 1, стадія C: до розчину метил-4-метил-3-оксовалерату (27,1 мл, 0,19 моль) в тетрагідрофурані при температурі 0 °C додають гідрид натрію (8,29 г, 0,21 моль, 60 % дисперсія в маслі). Через 30 хв витримки при кімнатній температурі додають розчин 4-бром-1-хлорметил2-метилбензолу (35,0 г, 0,16 моль) в тетрагідрофурані (50 мл). Одержану суміш гріють при температурі 70 °C протягом ночі (18 год). Додають 1,0 М розчин HCl (20 мл) для гасіння реакції. Екстрагують етилацетатом (200 мл), екстракт промивають водою (200 мл) і розсолом (200 мл), 6 UA 113086 C2 5 10 15 20 25 сушать над Na2SO4, фільтрують, і концентрують при зниженому тиску. Одержаний залишок розчиняють в толуолі (200 мл), і додають моногідрат гідразину (23,3 мл, 0,48 моль). Суміш гріють при температурі 120 °C протягом 2 год з насадкою Діна-Старка для видалення води. Суміш охолоджують, і розчинник видаляють при зниженому тиску, залишок розчиняють діхлорометаном (50 мл) і метанолом (50 мл). Цей розчин повільно виливають в хімічну склянку з водою (250 мл). Одержаний осаджений продукт збирають вакуумним фільтруванням. Сушать in vacuo в печі протягом ночі при температурі 40 °C, і одержують вказану в заголовку сполуку у вигляді твердої речовини (48,0 г, 0,16 моль). MS (m/z): 311,0 (M+1), 309,0 (M-1). Альтернативний синтез 4-[(4-бром-2-метилфеніл)метил]-5-ізопропіл-1Н-піразол-3-олу Одержують розчин 4-бром-1-хлорметил-2-метилбензолу (13,16 г, 59,95 ммоль) в ацетонітрилі (65,8 мл). Додають карбонат калію (24,86 г, 179,9 ммоль), йодид калію (11,94 г, 71,94 ммоль) і метил-4-метил-3-оксовалерат (8,96 мл; 62,95 ммоль). Одержану суміш перемішують при кімнатній температурі протягом 20 год. Додають соляну кислоту (2 н. розчин) до досягнення рівня рН 3. Розчин екстрагують етилацетатом (100 мл), органічну фазу промивають розсолом (100 мл), і сушать над Na 2SO4. Суміш фільтрують, і концентрують при зниженому тиску. Залишок розчиняють у толуолі (65,8 мл), і додають моногідрат гідразину (13,7 мл, 0,180 моль). Одержану суміш нагрівають зі зворотним холодильником, і воду видаляють із застосуванням насадки Діна-Старка. Через 3 год суміш охолоджують до температури 90 °C, додають додаткову кількість моногідрату гідразину (13,7 мл, 0,180 моль), і суміш нагрівають зі зворотним холодильником протягом 1 год. Суміш охолоджують, і концентрують при зниженому тиску. Одержану тверду речовину розтирають з водою (200 мл), фільтрують, і сушать у вакуумній печі над P2O5 при температурі 60 °C. Тверду речовину розтирають в ізогексані (200 мл), фільтрують, і одержують вказану в заголовку сполуку (14,3 г; вихід 77,1 %). MS (m/z): 309/311 (M+1). Препаративна методика 4 Синтез 4-(4-бром-2-метилбензил)-5-(пропан-2-іл)-1Н-піразол-3-іл-2,3,4,6-тетра-О-бензоїлбета-D-глюкопіранозиду 30 35 40 Схема 1, стадія D: в 1 л колбу додають 4-[(4-бром-2-метилфеніл)метил]-5-ізопропіл-1Нпіразол-3-ол (20 г, 64,7 ммоль), альфа-D-глюкопіранозилброміду тетрабензоат (50 г, 76 ммоль), бензилтрибутиламонію хлорид (6 г, 19,4 ммоль), дихлорометан (500 мл), карбонат калію (44,7 г, 323 ммоль) і воду (100 мл). Реакційну суміш перемішують протягом ночі при кімнатній температурі. Екстрагують дихлорометаном (500 мл). Екстракт промивають водою (300 мл) і розсолом (500 мл). Органічну фазу сушать над сульфатом натрію, фільтрують, і концентрують при зниженому тиску. Залишок очищають засобами флеш-хроматографії, і одержують вказану в заголовку сполуку (37 г, 64 ммоль). MS (m/z): 889,2 (M+1), 887,2 (M-1). Препаративна методика 5 Синтез 4-{4-[(1Е)-4-гідроксибут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2-іл)-1Н-піразол-3-іл2,3,4,6-тетра-О-бензоїл-бета-D-глюкопіранозиду 7 UA 113086 C2 5 10 Схема 1, стадія Е: до розчину 4-(4-бром-2-метилбензил)-5-(пропан-2-іл)-1H-піразол-3-іл2,3,4,6-тетра-О-бензоїл-бета-D-глюкопіранозиду (3 г, 3,4 ммоль) в ацетонітрилі (30 мл) і триетиламіні (20 мл) додають 3-бутен-1-ол (0,58 мл, 6,8 ммоль). Розчин знегажують азотом протягом 10 хв. Додають три-о-толілфосфін (205 мг, 0,67 ммоль) і ацетат паладію (76 мг, 0,34 ммоль). Гріють зі зворотним холодильником при температурі 90 °C протягом 2 год. Охолоджують до кімнатноїтемператури, і концентрують при зниженому тиску для видалення розчинника. Залишок очищають засобами флеш-хроматографії, і одержують вказану в заголовку сполуку (2,1 г, 2,4 ммоль). MS (m/z): 878,4 (M+1). Препаративна методика 6 Синтез 4-{4-[(1Е)-4-оксибут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2-іл)-1Н-піразол-3-іл-2,3,4,6тетра-О-бензоїл-бета-D-глюкопіранозиду 15 20 25 Схема 1, стадія F: до розчину 4-{4-[(1Е)-4-гідроксибут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2іл)-1Н-піразол-3-іл-2,3,4,6-тетра-О-бензоїл-бета-D-глюкопіранозиду (280 мг, 0,32 ммоль) і бікарбонату натрію (133,8 мг, 1,6 ммоль) в дихлорометані (20 мл) при температурі 0 °C додають 3,3,3-триацетокси-3-йодофталід (134 мг, 0,96 ммоль). Через 15 хв витримки при кімнатній температурі реакцію гасять насиченим водним розчином тіосульфату натрію (10 мл). Екстрагують дихлорометаном (30 мл). Екстракт промивають водою (30 мл) і розсолом (40 мл). Органічну фазу сушать над сульфатом натрію, фільтрують, і концентрують при зниженому тиску. Одержаний залишок очищають засобами флеш-хроматографії, і одержують вказану в заголовку сполуку (270 мг, 0,31 ммоль). MS (m/z): 876,5 (M+1), 874,5 (M-1). Препаративна методика 7 Синтез трет-бутил-2-{(3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6-тетра-O-бензоїл-бета-Dглюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл}-2,9-діазаспіро[5.5]ундекан-9карбоксилату 30 8 UA 113086 C2 5 10 Схема 1, стадія G: до розчину 4-{4-[(1Е)-4-оксибут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2-іл)1Н-піразол-3-іл-2,3,4,6-тетра-О-бензоїл-бета-D-глюкопіранозиду (270 мг, 0,31 ммоль) і гідрохлориду трет-бутил-2,9-діазаспіро[5.5]ундекан-9-карбоксилату (179 мг, 0,62 ммоль) в 1,2дихлороетані (5 мл) додають триацетоксиборогідрид натрію (98 мг, 0,46 ммоль). Через 30 хв витримки при кімнатній температурі реакцію гасять насиченим водним розчином бікарбонату натрію (10 мл). Екстрагують дихлорометаном (30 мл). Екстракт промивають водою (30 мл) і розсолом (40 мл), органічну фазу сушать над сульфатом натрію, фільтрують, і концентрують при зниженому тиску. Одержаний залишок очищають засобами флеш-хроматографії, і одержують вказану в заголовку сполуку (275 мг, 0,25 ммоль). MS (m/z): 1115,6 (M+1). Препаративна методика 8 Синтез 4-{4-[(1E)-4-(2,9-діазаспіро[5.5]ундец-2-іл)бут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2іл)-1H-піразол-3-іл-2,3,4,6-тетра-О-бензоїл-бета-D-глюкопіранозиду дигідрохлориду 15 2HCl 20 25 Схема 1, стадія Н: до розчину трет-бутил-2-{(3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6тетра-O-бензоїл-бета-D-глюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл}-2,9діазаспіро[5.5]ундекан-9-карбоксилату (275 мг, 0,25 ммоль) в дихлорометані (5 мл) додають хлористий водень (4,0 М розчин в 1,4-діоксані, 0,6 мл, 2,4 ммоль). Після витримування протягом ночі (18 год) при кімнатній температурі розчин концентрують при зниженому тиску для видалення розчинника, і одержують вказану в заголовку сполуку у вигляді твердої речовини (258 мг, 0,24 ммоль). MS (m/z): 1015,6 (M+1). Приклад 1 Синтез 4-{4-[(1E)-4-(2,9-діазаспіро[5.5]ундец-2-іл)бут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2іл)-1H-піразол-3-іл-бета-D-глюкопіранозиду 9 UA 113086 C2 5 10 Схема 1, стадія I: до розчину 4-{4-[(1E)-4-(2,9-діазаспіро[5.5]ундец-2-іл)бут-1-ен-1-іл]-2метилбензил}-5-(пропан-2-іл)-1H-піразол-3-іл-2,3,4,6-тетра-О-бензоїл-бета-D-глюкопіранозиду дигідрохлориду (258 мг, 0,24 ммоль) в метанолі (2 мл) додають гідроксид натрію (0,5 мл, 0,5 ммоль, 1,0 М розчин). Через 2 год витримки при температурі 40 °C розчин концентрують при зниженому тиску для видалення розчинника, і одержують залишок, який очищають засобами препаративної ВЕРХ: високий рівень рН, 25 % В протягом 4 хв, 25-40 % В протягом 4 хв при 85 мл/хв із застосуванням колонки C18XBridge ODB (3075 мм, 5 мкм), розчинник А Н2О+NH4HCO3, рН 10, розчинник В - MeCN, і одержують вказану в заголовку сполуку у вигляді твердої речовини (46 мг, 0,08 ммоль). MS (m/z): 598,8 (M+1), 596,8 (M-1). 10 UA 113086 C2 Схема 2 5 Препаративна методика 9 Синтез 4-(4-бром-2-метилбензил)-5-(пропан-2-іл)-1Н-піразол-3-іл-2,3,4,6-тетра-О-ацетилбета-D-глюкопіранозиду 11 UA 113086 C2 5 10 15 20 25 30 35 Схема 2, стадія А: в 1 л колбу додають 4-[(4-бром-2-метилфеніл)метил]-5-ізопропіл-1Нпіразол-3-ол (24 г, 77,6 ммоль), 2,3,4,6-тетра-О-ацетил-альфа-D-глюкопіранозилбромід (50,4 г, 116 ммоль), бензилтрибутиламонію хлорид (5 г, 15,5 ммоль), дихлорометан (250 мл), карбонат калію (32 г, 323 ммоль) і воду (120 мл). Реакційну суміш перемішують протягом ночі при кімнатній температурі. Екстрагують дихлорометаном (450 мл). Екстракт промивають водою (300 мл) і розсолом (500 мл). Органічну фазу сушать над сульфатом натрію, фільтрують, і концентрують при зниженому тиску. Одержаний залишок очищають засобами флешхроматографії, і одержують вказану в заголовку сполуку (36,5 г, 57 ммоль). MS (m/z): 638,5 (M+1), 636,5 (M-1). Альтернативний синтез 4-(4-бром-2-метилбензил)-5-(пропан-2-іл)-1Н-піразол-3-іл-2,3,4,6тетра-О-ацетил-бета-D-глюкопіранозиду Реагенти 4-[(4-бром-2-метилфеніл)метил]-5-ізопропіл-1Н-піразол-3-ол (24,0 г, 77,6 ммоль), 2,3,4,6-тетра-О-ацетил-альфа-D-глюкопіранозилбромід (50,4 г, 116 ммоль), бензилтрибутиламонію хлорид (4,94 г, 15,52 ммоль), карбонат калію (32,18 г, 232,9 ммоль), дихлорометан (250 мл) і воду (120 мл) об'єднують, і суміш перемішують при кімнатній температурі протягом 18 год. Суміш розподіляють між дихлорометаном (250 мл) і водою (250 мл). Органічну фазу промивають розсолом (250 мл), сушать над Na 2SO4, фільтрують, і концентрують при зниженому тиску. Одержаний залишок очищають засобами флешхроматографії (з елююванням градієнтом від 10 % до 70 % розчину етилацетату в дихлорометані), і одержують вказану в заголовку сполуку (36,5 г, вихід 74 %). MS (m/z): 639/641 (M+1). Препаративна методика 10 Синтез 4-{4-[(1Е)-4-гідроксибут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2-іл)-1Н-піразол-3-іл2,3,4,6-тетра-О-ацетил-бета-D-глюкопіранозиду Схема 2, стадія В: до розчину 4-(4-бром-2-метилбензил)-5-(пропан-2-іл)-1H-піразол-3-іл2,3,4,6-тетра-О-ацетил-бета-D-глюкопіранозиду (15 г, 23,5 ммоль) в ацетонітрилі (200 мл) і триетиламіні (50 мл) додають 3-бутен-1-ол (6,1 мл, 70 ммоль). Розчин знегажують азотом протягом 10 хв. Додають три-о-толілфосфін (1,43 г, 4,7 ммоль) й ацетат паладію (526 мг, 2,35 ммоль). Після кип'ятіння зі зворотним холодильником при температурі 90 °C протягом 2 год суміш охолоджують, і концентрують при зниженому тиску для видалення розчинника. Одержаний залишок очищають засобами флеш-хроматографії, і одержують вказану в заголовку сполуку (7,5 г, 11,9 ммоль). MS (m/z): 631,2 (M+1), 629,2 (M-1). Препаративна методика 11 12 UA 113086 C2 Синтез 4-{4-[(1Е)-4-оксибут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2-іл)-1Н-піразол-3-іл-2,3,4,6тетра-О-ацетил-бета-D-глюкопіранозиду 5 10 15 Схема 2, стадія С: до розчину 4-{4-[(1Е)-4-гідроксибут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2іл)-1Н-піразол-3-іл-2,3,4,6-тетра-О-ацетил-бета-D-глюкопіранозиду (1,5 г, 2,38 ммоль) і бікарбонату натрію (2 г, 23,8 ммоль) в дихлорометані (50 мл) при температурі 0 °C додають 3,3,3-триацетокси-3-йодофталід (2,1 г, 4,76 ммоль). Через 15 хв витримки при кімнатній температурі реакцію гасять насиченим водним розчином тіосульфату натрію (10 мл). Екстрагують дихлорометаном (30 мл), екстракт промивають водою (30 мл) і розсолом (40 мл). Органічну фазу сушать над сульфатом натрію, фільтрують, і концентрують при зниженому тиску. Одержаний залишок очищають засобами флеш-хроматографії, і одержують вказану в заголовку сполуку (0,95 г, 1,51 ммоль). MS (m/z): 628,8(M+1), 626,8 (M-1). Препаративна методика 12 Синтез трет-бутил-2-{(3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6-тетра-O-ацетил-бета-Dглюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл}-2,9-діазаспіро[5.5]ундекан-9карбоксилату 20 25 30 35 Схема 2, Стадія D: до розчину 4-{4-[(1Е)-4-оксибут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2-іл)1Н-піразол-3-іл-2,3,4,6-тетра-О-ацетил-бета-D-глюкопіранозиду (600 мг, 0,95 ммоль) і третбутил-2,9-діазаспіро[5.5]ундекан-9-карбоксилату гідрохлориду (333 мг, 1,2 ммоль) в 1,2дихлороетані (30 мл) додають триацетоксиборогідрид натрію (303 мг, 1,4 ммоль). Через 30 хв витримки при кімнатній температурі реакцію гасять насиченим водним розчином бікарбонату натрію (15 мл). Розчин екстрагують дихлорометаном (60 мл). Екстракт промивають водою (30 мл) і розсолом (60 мл). Органічну фазу сушать над сульфатом натрію, фільтрують, і концентрують при зниженому тиску. Одержаний залишок очищають засобами флешхроматографії, і одержують вказану в заголовку сполуку (500 мг, 0,58 ммоль). MS (m/z): 866,8, 867,8 (M+1), 864,8, 865,8 (M-1). Препаративна методика 13 Синтез трет-бутил-2-{(3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6-тетра-O-ацетил-бета-Dглюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл}-2,8-діазаспіро[4.5]декан-8карбоксилату 13 UA 113086 C2 5 Указану в заголовку сполуку одержують по суті за методом, наведеним у Препаративній методиці 12. MS (m/z): 852,8, 853,6 (M+1), 850,8, 851,6 (M-1). Препаративна методика 14 Синтез трет-бутил-9-{(3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6-тетра-O-ацетил-бета-Dглюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл}-3,9-діазаспіро[5.5]ундекан-3карбоксилату 10 15 Указану в заголовку сполуку одержують по суті за методом, наведеним у Препаративній методиці 12. MS (m/z): 866,8, 867,6 (M+1), 864,8, 865,6 (M-1). Препаративна методика 15 Синтез 4-{4-[(1E)-4-(2,9-діазаспіро[5.5]ундец-2-іл)бут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2іл)-1H-піразол-3-іл-2,3,4,6-тетра-О-ацетил-бета-D-глюкопіранозиду дигідрохлориду 2HCl 20 Схема 2, стадія Е: до розчину трет-бутил-2-{(3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6тетра-O-ацетил-бета-D-глюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл}-2,9діазаспіро[5.5]ундекан-3-карбоксилату (500 мг, 0,58 ммоль) в дихлорометані (20 мл) додають хлористий водень (4,0 М розчин в 1,4-діоксані, 1,5 мл, 5,8 ммоль). Після 2 год витримки при 14 UA 113086 C2 5 кімнатній температурі розчин концентрують при зниженому тиску для видалення розчинника, і одержують вказану в заголовку сполуку у вигляді твердої речовини (480 мг, 0,57 ммоль). MS (m/z): 767,4 (M+1). Препаративна методика 16 Синтез 4-{4-[(1E)-4-(2,8-діазаспіро[4.5]дец-2-іл)бут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2-іл)1H-піразол-3-іл-2,3,4,6-тетра-О-ацетил-бета-D-глюкопіранозиду дигідрохлориду 2HCl 10 Указану в заголовку сполуку одержують по суті за методом, наведеним у Препаративній методиці 15. MS (m/z): 752,8, 753,8 (M+1), 750,8 (M-1). Перший альтернативний синтез сполуки Прикладу 1 Перший альтернативний синтез 4-{4-[(1E)-4-(2,9-діазаспіро[5.5]ундец-2-іл)бут-1-ен-1-іл]-2метилбензил}-5-(пропан-2-іл)-1H-піразол-3-іл-бета-D-глюкопіранозиду 15 20 25 30 Схема 2, стадія F: до 4-{4-[(1E)-4-(2,9-діазаспіро[5.5]ундец-2-іл)бут-1-ен-1-іл]-2метилбензил}-5-(пропан-2-іл)-1H-піразол-3-іл-2,3,4,6-тетра-О-ацетил-бета-D-глюкопіранозиду дигідрохлориду (480 мг, 0,24 ммоль) додають метанол (5 мл), триетиламін (3 мл) і воду (3 мл). Після витримки протягом ночі (18 год) при кімнатній температурі розчин концентрують при зниженому тиску до суха. Одержаний залишок очищають засобами препаративної ВЕРХ: високий рівень рН, 25 % В протягом 4 хв, 25-40 % В протягом 4 хв при 85 мл/хв із застосуванням колонки C18XBridge ODB (3075 мм, 5 мкм), розчинник А - Н2О+NH4HCO3, рН 10, розчинник В MeCN, і одержують вказану в заголовку сполуку у вигляді твердої речовини (50 мг, 0,08 ммоль). 1 MS (m/z): 598,8 (M+1), 596,8 (M-1). H ЯМР (400,31 МГц, CD3OD): δ 7,11 (d, J=1,3 Гц, 1H), 7,04 (dd, J=1,3 Гц, 8,0 Гц, 1H), 6,87 (d, J=8,0 Гц, 1H), 6,36 (d, J=15,8 Гц, 1H), 6,16 (dt, J=15,8 Гц, 6,3 Гц, 1H), 5,02 (m, 1H), 3,81 (d, J=11,7 Гц, 1H), 3,72 (d, J=16,8 Гц, 1H), 3,68 (d, J=16,8 Гц, 1H), 3,64 (m, 1H), 3,37-3,29 (m, 4H), 2,79 (m, 1H), 2,72 (t, J=5,8 Гц, 4H), 2,44-2,33 (m, 6H), 2,30 (s, 3H), 2,26 (широкий s, 2H), 1,59 (m, 2H), 1,50 (m, 2H), 1,43 (m, 2H), 1,36 (m, 2H), 1,11 (d, J=7,0 Гц, 3H), 1,10 (d, J=7,0 Гц, 3H). Приклад 2 Синтез 4-{4-[(1E)-4-(2,8-діазаспіро[4.5]дец-2-іл)бут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2-іл)1H-піразол-3-іл-бета-D-глюкопіранозиду 35 15 UA 113086 C2 5 10 Указану в заголовку сполуку одержують по суті за методом, наведеним у Першому альтернативному синтезі сполуки Прикладу 1. MS (m/z): 584,7 (M+1), 582,8 (M-1). Приклад 3 Синтез 4-{4-[(1E)-4-(3,9-діазаспіро[5.5]ундец-3-іл)бут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2іл)-1H-піразол-3-іл-бета-D-глюкопіранозиду По суті, для одержання вказаної в заголовку сполуки спочатку сполуку Препаративної методики 14 обробляють хлористим воднем, як описано в Препаративній методиці 15, потім одержану гідрохлоридну сіль обробляють триетиламіном, як описано в Першому альтернативному синтезі сполуки Прикладу 1. MS (m/z): 598,8, 599,8 (M+1), 596,8, 597,8 (M-1). 16 UA 113086 C2 Схема 3 5 Препаративна методика 17 Синтез трет-бутил-4-бут-3-ініл-4,9-діазаспіро[5.5]ундекан-9-карбоксилату 17 UA 113086 C2 5 10 Схема 3, стадія A: до суспензії гідрохлориду трет-бутил-4,9-діазаспіро-[5.5]ундекан-9карбоксилату (16,66 г, 57,28 ммоль) в ацетонітрилі (167 мл) додають карбонат цезію (46,66 г, 143,21 ммоль). Одержану суміш перемішують протягом 10 хв при температурі навколишнього середовища, після чого додають 4-бромбутин (6,45 мл, 68,74 ммоль). Реакційну суміш нагрівають зі зворотним холодильником, і перемішують протягом 18 год. Суміш охолоджують, і концентрують при зниженому тиску. Залишок розподіляють між водою (200 мл) і етилацетатом (150 мл). Фази відокремлюють, і водний шар екстрагують етилацетатом (100 мл). Об'єднані органічні шари промивають водою (200 мл), потім розсолом (150 мл), сушать над MgSO 4, фільтрують, концентрують при зниженому тиску, і одержують вказану в заголовку сполуку (17,2 1 г, вихід 98 %). H ЯМР (300,11 МГц, CDCl3): δ 3,43-3,31 (m, 4H), 2,53-2,48 (m, 2H), 2,37-2,29 (m, 4H), 2,20 (s, 2H), 1,94 (t, J=2,6 Гц, 1H), 1,44 (s, 17H). Препаративна методика 18 Синтез трет-бутил-4-[(E)-4-(4,4,5,5-тетраметил-1,3,2-діоксаборолан-2-іл)бут-3-еніл]-4,9діазаспіро[5.5]ундекан-9-карбоксилату 15 20 25 Схема 3, стадія В: до трет-бутил-4-бут-3-ініл-4,9-діазаспіро[5.5]ундекан-9-карбоксилату (17,21 г, 56,16 ммоль) додають триетиламін (5,62 ммоль; 0,783 мл), 4,4,5,5-тетраметил-1,3,2діоксаборолан (8,56 мл, 59,0 ммоль) і хлорид цирконоцену (1,45 г, 5,62 ммоль). Одержану суміш нагрівають до температури 65 °C протягом 3,5 год. Суміш охолоджують, і розчиняють в дихлорометані (150 мл). Одержаний розчин пропускають через шар силікагелю завтовшки приблизно 4 см, і елююють дихлорометаном (2200 мл). Фільтрат концентрують при зниженому 1 тиску, і одержують вказану в заголовку сполуку (21,2 г, вихід 87 %). H ЯМР (300,11 МГц, CDCl3): δ 6,65-6,55 (m, 1H), 5,49-5,43 (m, 1H), 3,42-3,29 (m, 4H), 2,40-2,27 (m, 6H), 2,25-2,08 (m, 2H), 1,701,13 (m, 29H). Препаративна методика 19 Синтез трет-бутил-2-{(3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-бета-D-глюкопіранозил)окси]-1Hпіразол-4-іл}метил)феніл]бут-3-ен-1-іл}-2,9-діазаспіро[5.5]ундекан-9-карбоксилату 30 35 40 Схема 3, стадія С: розчин 4-(4-бром-2-метилбензил)-5-(пропан-2-іл)-1Н-піразол-3-іл-2,3,4,6тетра-О-ацетил-бета-D-глюкопіранозиду (20 г, 31,3 ммоль), трет-бутил-4-[(E)-4-(4,4,5,5тетраметил-1,3,2-діоксаборолан-2-іл)бут-3-еніл]-4,9-діазаспіро[5.5]ундекан-9-карбоксилату (16,3 г, 37,5 ммоль) і карбонату калію (12,97 г, 93,82 ммоль) в тетрагідрофурані (200 мл) і воді (40 мл) знегажують протягом 15 хв барботуванням азотом. Додають Pd(OAc) 2 (140 мг, 625 мкмоль) й 2дициклогексилфосфіно-2',4',6'-три-ізопропіл-1,1'-дифеніл (0,596 г, 1,25 ммоль), і реакційну суміш нагрівають зі зворотним холодильником протягом 16 год. Розчин охолоджують до температури навколишнього середовища, і додають метанол (200 мл). Через 30 хв розчинник видаляють при 18 UA 113086 C2 5 зниженому тиску. Суміш розподіляють між етилацетатом (500 мл) і розсолом (500 мл), додаючи при цьому водний розчин MgSO4 (1 М, 500 мл) для сприяння поділу фаз. Шари розділяють, органічний шар сушать над MgSO4, фільтрують через 10 см шар силікагелю, й елююють етилацетатом (приблизно 1,5 л). Фільтрат відкидають, а шар діоксиду кремнію промивають 5 % розчином МеОН в тетрагідрофурані (2 л). Метанольний фільтрат концентрують при зниженому тиску, і одержують вказану в заголовку сполуку (20,1 г, 92 %). Другий альтернативний синтез сполуки Прикладу 1 Другий альтернативний синтез 4-{4-[(1E)-4-(2,9-діазаспіро[5.5]ундец-2-іл)бут-1-ен-1-іл]-2метилбензил}-5-(пропан-2-іл)-1H-піразол-3-іл-бета-D-глюкопіранозиду 10 15 20 Схема 3, стадія D: до охолодженого льодяною водою розчину трет-бутил-2-{(3E)-4-[3-метил4-({5-(пропан-2-іл)-3-бета-D-глюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл}2,9-діазаспіро[5.5]ундекан-9-карбоксилату (14,87 г, 21,28 ммоль) в дихлорометані (149 мл) додають трифторооцтову кислоту (32,2 мл, 0,426 моль). Розчин витримують для нагрівання до кімнатної температури. Через 30 хв суміш повільно додають до розчину аміаку в метанолі (2 М, 300 мл), застосовуючи за необхідності охолодження для підтримання незмінної температури. Розчин перемішують при кімнатній температурі протягом 15 хв. Суміш концентрують при зниженому тиску, і залишок очищають із використанням смоли SCX-2. Основний фільтрат концентрують при зниженому тиску, і залишок розтирають/обробляють ультразвуком в етилацетаті, фільтрують, і сушать. Одержану тверду речовину розчиняють в МеОН (200 мл), і концентрують in vacuo. Це повторюють декілька разів, і одержують вказану в заголовку сполуку 20 (12,22 г, вихід 96 %). MS (m/z): 599 (M+1). [α]D =-12° (C=0,2, MeOH). 25 19 UA 113086 C2 Схема 4 5 Препаративна методика 20 Синтез (3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6-тетра-O-ацетил-бета-Dглюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл-метансульфонату 20 UA 113086 C2 5 10 Схема 4, стадія A: до розчину 4-{4-[(1Е)-4-гідроксибут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2іл)-1Н-піразол-3-іл-2,3,4,6-тетра-О-ацетил-бета-D-глюкопіранозиду (3,7, 5,87 ммоль) в дихлорометані (15 мл) і триетиламіні (4 мл, 29 ммоль) при температурі 0 °C. Після кип'ятіння зі зворотним холодильником при кімнатній температурі протягом 30 хв розчин концентрують при зниженому тиску для видалення розчинника. Залишок очищають засобами флешхроматографії, і одержують вказану в заголовку сполуку (2,9 г, 4,1 ммоль). MS (m/z): 708,5 (M+1), 706,5 (M-1). Препаративна методика 21 Синтез трет-бутил-2-{(3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6-тетра-O-ацетил-бета-Dглюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл}-2,6-діазаспіро[3.5]нонан-6карбоксилату 15 20 25 Схема 4, стадія В: до розчину (3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6-тетра-O-ацетилбета-D-глюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл-метансульфонату (200 мг, 0,28 ммоль) і трет-бутил-2,6-діазаспіро[3.5]нонан-6-карбоксилату (77 мг, 0,34 ммоль) в ацетонітрилі (3 мл) додають діізопропілетиламін (0,2 мл, 1,1 ммоль). Суміш гріють при температурі 80 °C протягом ночі. Концентрують при зниженому тиску, залишок очищають засобами флеш-хроматографії, і одержують вказану в заголовку сполуку (127 мг, 0,15 ммоль). MS (m/z): 838,8, 839,6 (M+1), 836,8, 837,6 (M-1). Препаративна методика 22 Синтез трет-бутил-2-{(3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6-тетра-O-ацетил-бета-Dглюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл}-2,7-діазаспіро[3.5]нонан-7карбоксилату 21 UA 113086 C2 5 Указану в заголовку сполуку одержують по суті за методом, наведеним у Препаративній методиці 21. MS (m/z): 838,8, 839,6 (M+1), 836,8, 837,6 (M-1). Препаративна методика 23 Синтез трет-бутил-7-{(3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6-тетра-O-ацетил-бета-Dглюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл}-2,7-діазаспіро[3.5]нонан-2карбоксилату 10 15 Указану в заголовку сполуку одержують по суті за методом, наведеним у Препаративній методиці 21. MS (m/z): 838,8, 839,6 (M+1), 836,8, 837,6 (M-1). Препаративна методика 24 Синтез трет-бутил-1-{(3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6-тетра-O-ацетил-бета-Dглюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл}-1,8-діазаспіро[4.5]декан-8карбоксилату 20 25 Указану в заголовку сполуку одержують по суті за методом, наведеним у Препаративній методиці 21. MS (m/z): 852,8, 853,6 (M+1), 850,8, 852,8 (M-1). Препаративна методика 25 Синтез трет-бутил-8-{(3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6-тетра-O-ацетил-бета-Dглюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]бут-3-ен-1-іл}-2,8-діазаспіро[4.5]декан-2 22 UA 113086 C2 карбоксилату 5 Указану в заголовку сполуку одержують по суті за методом, наведеним у Препаративній методиці 21. MS (m/z): 852,8, 853,6 (M+1), 850,8, 851,6 (M-1). Приклад 4 Синтез 4-{4-[(1E)-4-(2,6-діазаспіро[3.5]нон-2-іл)бут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2-іл)1H-піразол-3-іл-бета-D-глюкопіранозиду 10 15 20 25 Схема 4, стадія С: до розчину трет-бутил-2-{(3E)-4-[3-метил-4-({5-(пропан-2-іл)-3-[(2,3,4,6тетра-O-ацетил-бета-D-глюкопіранозил)окси]-1H-піразол-4-іл}метил)феніл]-бут-3-ен-1-іл}-2,6діазаспіро[3.5]нонан-6-карбоксилату в дихлорометані (2 мл) додають 4,0 М розчин HCl/1,4діоксан (1,5 мл, 1,5 ммоль), і перемішують при кімнатній температурі протягом 4,0 год. Суміш концентрують при зниженому тиску до одержання піноподібної твердої речовини. Цю тверду речовину протягом ночі обробляють 2,0 М розчином аміаку в MeOH (2 мл). Через 18 год витримки при кімнатній температурі розчин концентрують при зниженому тиску для видалення розчинника. Одержаний залишок очищають засобами препаративної ВЕРХ: високе значення рН, 19 % В протягом 3 хв, 19-34 % В протягом 5 хв при 85 мл/хв із застосуванням колонки C18XBridge ODB (3075 мм, 5 мкм), розчинник А - Н2О+NH4HCO3, рН 10, розчинник В - MeCN, і одержують вказану в заголовку сполуку у вигляді твердої речовини (47 мг, 0,08 ммоль). MS (m/z): 570,8, 571,8 (M+1), 568,7, 569,8 (M-1). Приклад 5 Синтез 4-{4-[(1E)-4-(2,7-діазаспіро[3.5]нон-2-іл)бут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2-іл)1H-піразол-3-іл-бета-D-глюкопіранозиду 30 23 UA 113086 C2 5 10 15 20 Указану в заголовку сполуку одержують по суті за методом, наведеним у Прикладі 4. MS (m/z): 570,8, 571,8 (M+1), 568,7, 569,8 (M-1). Приклад 6 Синтез 4-{4-[(1E)-4-(2,7-діазаспіро[3.5]нон-7-іл)бут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2-іл)1H-піразол-3-іл-бета-D-глюкопіранозиду трифтороацетату (1:2) Указану в заголовку сполуку одержують по суті за методом, наведеним у Прикладі 4, з очищенням кінцевої сполуки засобами препаративної ВЕРХ з низьким значенням рН (низьке значення рН, 16 % В протягом 3 хв, 16-33 % В протягом 5 хв при 85 мл/хв із застосуванням колонки C18XBridge ODB (3075 мм, 5 мкм), розчинник А - Н2О+0,1 % TFA, розчинник В MeCN+0,1 % TFA). MS (m/z): 570,8, 571,8 (M+1), 568,7, 569,8 (M-1). Приклад 7 Синтез 4-{4-[(1E)-4-(1,8-діазаспіро[4.5]дец-1-іл)бут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2-іл)1H-піразол-3-іл-бета-D-глюкопіранозиду Указану в заголовку сполуку одержують по суті за методом, наведеним у Прикладі 4. MS (m/z): 584,7, 585,8 (M+1), 582,8, 583,8 (M-1). Приклад 8 Синтез 4-{4-[(1E)-4-(2,8-діазаспіро[4.5]дец-8-іл)бут-1-ен-1-іл]-2-метилбензил}-5-(пропан-2-іл)1H-піразол-3-іл-бета-D-глюкопіранозиду 25 30 Указану в заголовку сполуку одержують по суті за методом, наведеним у Прикладі 4. MS (m/z): 584,7, 585,8 (M+1), 582,8, 583,8 (M-1). Дослідження натрій-залежного транспортера глюкози 1 (SGLT1) і SGLT2 кДНК, які кодують людський SGLT1 (slc5a1, NM_000343), людський SGLT2 (slc5a2, NM_003041) і мишачий SGLT1 (slc5a1, NM_019810.4), одержують від компаній Opеnbiosystems, 24 UA 113086 C2 5 10 15 20 25 Invitrogеn і Opеnbiosystems, відповідно. Ці кДНК клонують в pcDNA3.1+ для експресії ссавцями, і стабільно трансфекують в клітини яєчника китайського хом'ячка (СНО)-K1 стандартними методами трансфекування ссавців. SGLT-експресуючий субклон кожної надекспресуючої лінії клітин вибирають на основі стійкості до неоміцину (генетицин (Geneticine), компанія Invitrogеn) і 14 14 активності в дослідженні поглинання C-α-метил-D-глюкопіранозиду ( C-AMG) (дивись нижче). Стабільні SGLT-експресуючі клітини підтримують стандартними методами культивування клітин. 14 Активність SGLT визначають як натрій-залежне поглинання C-AMG у вищезгаданих клітинних лінях, як описано нижче. У кожну лунку 96-лункового планшета BioCoat poly-D-lysine (компанія Becton Dickinson) висівають 100 мкл культурального середовища, яке містить 30000 клітин, і культивують його при температурі 37 °C протягом ночі. Це культуральне середовище видаляють, і клітини двічі промивають 200 мкл реакційного буфера (140 мМ NaCl, 2 мМ KCl, 1 мМ CaCl2, MgCl2 і 14 мМ N-2-гідроетилпіперазин-N'-2-етансульфонової кислоти (HEPES), рН 7,5). Надлишок буфера видаляють паперовими серветками. У кожну лунку додають 35 мкл реакційного буфера. У кожну лунку додають 5 мкл 10 % розчину диметилсульфоксиду (DMSO) в реакційному буфері, який містить різні концентрації досліджуваної сполуки або як контроль не 14 містить сполуки. Реакцію ініціюють додаванням 10 мкл C-AMG в реакційному буфері для доведення кінцевої концентрації до 4 мкM. Планшет інкубують при температурі 37 °C протягом 125 хв. Реакцію зупиняють аспіруванням реакційного буфера з подальшим потрійним промиванням 200 мкл реакційного буфера, що має температуру танення льоду. Для забезпечення повного видалення реакційного буфера вдаються до ручної аспірації. У кожну лунку додають спочатку 10 мкл 0,1 н. розчину NaOH, потім 100 мкл сцинтиляційного коктейлю Supermix (компанія PerkinElmer). Після змішування сцинтиляційний сигнал планшета аналізують лічильником MicroBeta (компанія PerkinElmer). Десятидозову реакційну криву підганяють до емпіричної чотирипараметричної моделі із використанням ActivityBase (компанія ID Business Solution) для визначення концентрації інгібітора при напівмаксимальному інгібуванні (IC50). Сполуки Прикладів 1-8 за цим винаходом випробовують по суті як описано вище, і вони демонструють значення IC50 для SGLT1 нижче ніж приблизно 500 нМ. Таблиця 1 In vitro активність сполуки Прикладу 1 проти SGLT1 і SGLT2 Експериментальна сполука Приклад 1 Людський SGLT1 IC50, нМ 26±20 (n=10) Людський SGLT2 IC50, Мишачий SGLT1 IC50, нМ нМ 6100±1200 (n=10) 10±2 (n=9) 30 35 40 45 50 Отже, дані в Таблиці 1 свідчать, що сполука Прикладу 1 інгібує людський і мишачий SGLT1 in vitro, і є більш активною in vitro проти людського і мишачого SGLT1, аніж проти людського SGLT2. Глюкозознижувальні ефекти при проведенні перорального глюкозотолерантного випробування (OGTT) Досліджувану сполуку одержують доданням до попередньо зваженої досліджуваної сполуки ® носія, який містить 1 % гідроксіетилцелюлози, 0,25 % Tween 80 з 0,05 % піногасника, для одержання 1 мг/мл розчину. Суміш обробляють ультразвуком протягом приблизно 30 с. Одержаний розчин використовують як вихідний розчин, з якого за рахунок розбавлення носієм одержують дозовані розчини нижчої концентрації. Індивідуально розміщені миші лінії C57Bl/6 голодують протягом ночі внаслідок припинення доступу до їжі пізно ввечері напередодні дня випробування. Наступного ранку мишей зважують, і надрізанням хвостової вени відбирають одну пробу крові натщесерце для визначення рівня глюкози глюкометром (глюкометр AccuChek, компанія Roche). Експериментальні групи (n=5) визначають на підставі рівня глюкози в крові натщесерце, і за варіантом, якому віддають перевагу, ці групи включають тварин з рівнем глюкози в діапазоні 80-100 мг/дл. Після розподілення на групи першій миші перорально шлунковим зондом вводять препарат експериментальної сполуки в дозі 10 мл на кг маси тіла, і запускають таймер. Кожній наступній тварині дозу вводять з інтервалом у півтори хвилини. Через три години після початку першої обробки сполукою відбирають базову пробу крові для визначення рівня глюкози (від першої тварини, через надріз хвостової вени). Негайно після цього тварині перорально вводять дозу 50 % декстрози (компанія Hospira) із розрахунку 3 г на кг маси тіла. Проби крові на глюкозу відбирають з хвостової вени точно з півторахвилинним інтервалом, Отже, кров від кожної 25 UA 113086 C2 тварини збирають через 20, 40, 60 і 120 хвилин після дози декстрози. Таблиця 2 Глюкозознижувальні ефекти при проведенні OGTT Результати перорального глюкозотолерантного тесту Середнє±середня квадратична похибка Двофакторний дисперсійний аналіз/метод Бонфероні *p
ДивитисяДодаткова інформація
Автори англійськоюQu, Fucheng, Mantlo, Nathan Bryan
Автори російськоюЦюй Фучен, Ментлоу Нейтан Браян
МПК / Мітки
МПК: C07D 471/10, A61K 31/4155, C07D 487/10, A61K 31/438
Мітки: піразольні, sglt1, сполуки, інгібітори
Код посилання
<a href="https://ua.patents.su/31-113086-pirazolni-spoluki-yak-ingibitori-sglt1.html" target="_blank" rel="follow" title="База патентів України">Піразольні сполуки як інгібітори sglt1</a>
Піразольні сполуки для боротьби з безхребетними шкідниками

Номер патенту: 103006
Опубліковано: 10.09.2013
Автори: Кьорбер Карстен, Пуль Міхаель, Гросс Штеффен, фон Дейн Вольфганг, Бастіаанс Хенрікус Маріа Мартінус, Анспо Дуглас Д., Бройнінгер Делфін, Калбертсон Дебора Л., Олоумі-Садегі Хассан
МПК: A01N 47/18, A01N 43/74, C07D 419/00, A01N 43/707, A01N 43/56, A01P 7/04
Мітки: сполуки, піразольні, боротьби, шкідниками, безхребетними
Формула / Реферат:
1. Спосіб боротьби з безхребетними шкідниками, вибраними з комах, павукоподібних і нематод, який включає обробку шкідників, їх харчових ресурсів, їх місця поширення або їх місця розмноження, або рослин, насіння, ґрунту, ділянки, матеріалу, або навколишнього середовища, у якому шкідники ростуть або можуть рости, або матеріалів, рослин, насіння, ґрунтів, поверхонь, або просторів, які підлягають захисту від нападу або інвазії шкідниками,...
Піразольні сполуки для боротьби з безхребетними шкідниками

Номер патенту: 103633
Опубліковано: 11.11.2013
Автори: Польман Маттіас, Кьорбер Карстен, Анспо Дуглас Д., Олоумі-Садегі Хассан (покійний), Калбертсон Дебора Л., Дешмукх Прашант, Кайзер Флоріан, Дікхаут Йоахім, ле Везуе Ронан, фон Дейн Вольфганг, Гросс Штеффен, Зьоргель Себастіан
МПК: C07D 403/12, A01N 43/56
Мітки: боротьби, піразольні, сполуки, шкідниками, безхребетними
Формула / Реферат:
1. Піразольні сполуки формул І або II та їх солі й N-оксиди, (I) , (II)деА означає піразольний радикал формул А1, А2 або A3 , A1 ,...
Дициклічні гетероароматичні сполуки як інгібітори білкової тирозинкінази

Номер патенту: 66827
Опубліковано: 15.06.2004
Автори: антріп Стівен Беррі, Картер Малколм Клайв, Лекі Керін Елізабет, Сміт Катрін Джейн, Кокерілл Джордж Стюарт
МПК: C07D 471/04, A61K 31/4709, A61P 35/00, A61P 17/06, A61K 31/517, C07D 239/94, C07D 417/14, C07D 405/04, C07D 405/14, A61P 29/00, A61K 31/519, A61P 43/00, C07D 417/04
Мітки: інгібітори, сполуки, білкової, дициклічні, гетероароматичні, тирозинкінази
Формула / Реферат:
1. Дициклічні гетероароматичні сполуки формули (І): (І)або їх солі чи сольвати як інгібітори білкової тирозинкінази,деY - CR1, a V - N,або Y - CR1, a V - CR2,R1 - група CH3SO2CH2CH2NHCH2-Ar, причому Аr вибрано з групи, яка складається з фурану та тіазолу, кожний з яких заміщено, як варіант, одним чи двома галогенами,...
Піримідини як інгібітори сорбітдегідрогенази, фармацевтична композиція, що їх містить, проміжні сполуки та спосіб одержання проміжної сполуки

Номер патенту: 71951
Опубліковано: 17.01.2005
Автори: Зембровскі Уільям Джеймс, Чу-Моєр Маргарет Юхуа, Маррі Джеррі Ентоні, Міларі Банавара Лакшман
МПК: C07D 487/08, C07D 409/14, C07D 491/048, A61P 9/10, C07D 451/02, C07D 451/06, C07D 403/14, A61K 31/5377, A61K 31/517, C07D 491/20, A61P 3/10, A61K 31/506, C07D 521/00, C07D 491/04, C07D 401/14, C07D 417/14, C07D 405/14, C07D 491/10, C07D 513/10, C07D 498/04, C07D 401/12, C07D 471/08, C07D 403/04, C07D 239/42, C07D 405/12, C07D 487/04, C07D 413/12, C07D 417/12, C07D 409/12, C07D 471/04, C07D 403/12, A61K 31/53, C07D 401/04, A61P 43/00
Мітки: проміжні, сорбітдегідрогенази, спосіб, фармацевтична, піримідини, інгібітори, одержання, сполуки, композиція, проміжної, містить
Формула / Реферат:
1. Похідне піримідину формули I: , Iйого пролікарська форма або фармацевтично прийнятна сіль згаданої сполуки або згаданої пролікарської форми, де: R1 є (R)-1-гідроксіетилом; R2 є воднем; R3 є; іR9 є піримідилом або триазинілом; згаданий піримідил або...
Сполуки, ефективні як інгібітори ксантиноксидази, спосіб одержання таких сполук (варіанти) та фармацевтична композиція, що містить такі сполуки

Номер патенту: 110197
Опубліковано: 10.12.2015
Автори: Йунг Чол Кю, Коо Кі Чул, Парк Док Сонг, Парк Хюн Йюнг, Кім Гин Тае, Чоі Сунг Піл, Парк Ван Су, Артемов Василій, Чоі Ин Сіл, Кім Тае Хун, Сонг Йонг Ук, Парк Хеуі Сул
МПК: A61P 3/10, C07D 209/04, A61K 31/40, C07D 403/02
Мітки: містить, ефективні, таких, спосіб, ксантиноксидази, сполук, інгібітори, одержання, варіанти, сполуки, такі, фармацевтична, композиція
Формула / Реферат:
1. Сполуки наступної формули (1):, (1) в якій: А вибирається з наступних заміщень А-і, A-iv, A-v, A-vi та A-vii: , ,
Попередній патент: Вакуум-віброекстрактор періодичної дії з комбінованим енергопідведенням
Наступний патент: Природна холодна поверхнева обробка порожнистих скляних виробів
Випадковий патент: Вентиляційна решітка