Феноксіетилпіперидинові сполуки







Формула / Реферат
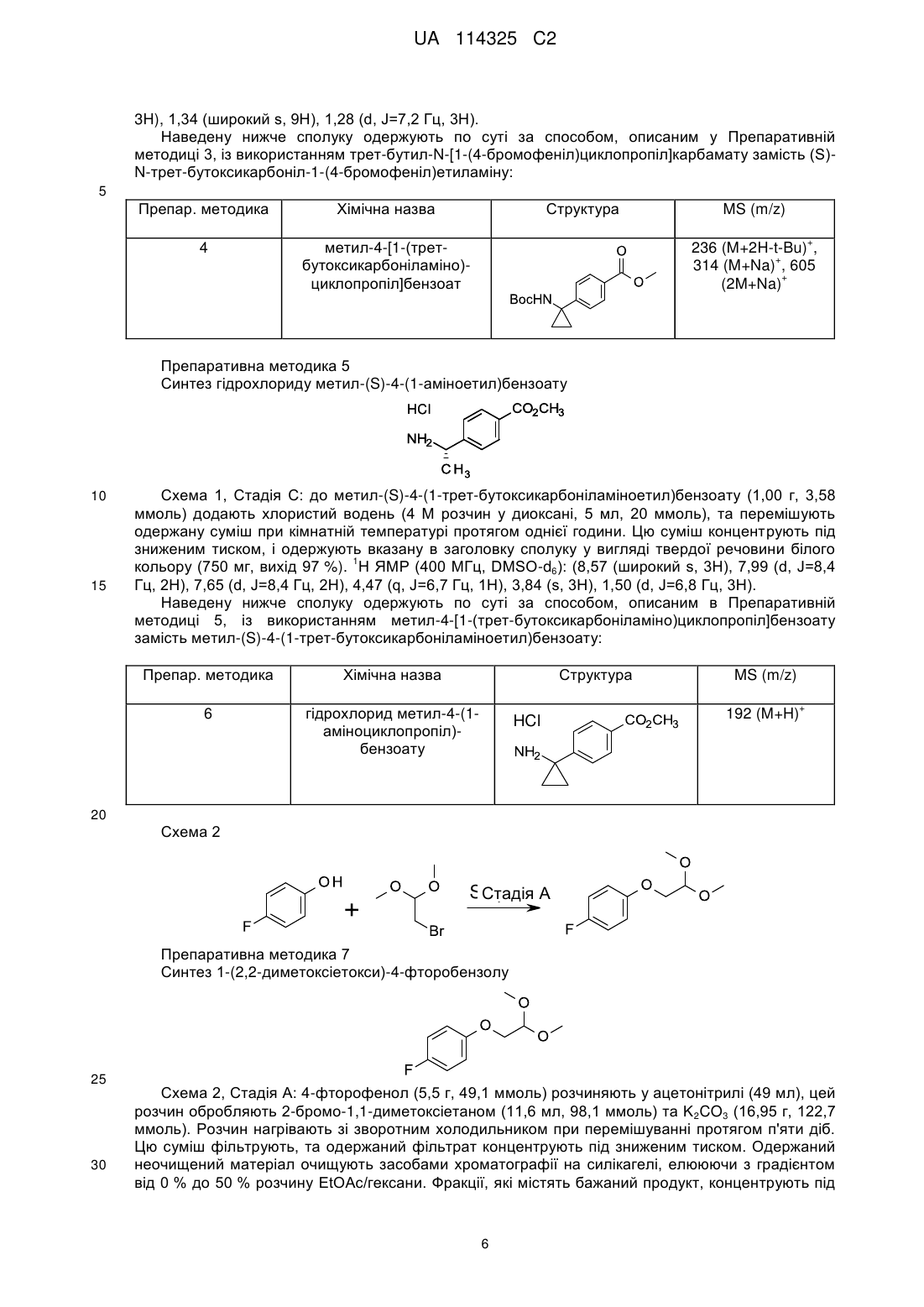
1. Сполука формули:
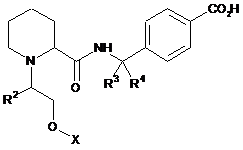 ,
,
де X являє собою:
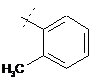 ,
,  або
або 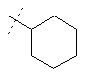 ,
,
R1 являє собою Н, -CN або F;
R2 являє собою Н або метил;
R3 являє собою Н; та
R4 являє собою Н, метил або етил; або
R3 та R4, сполучені разом, утворюють циклопропілове кільце;
або її фармацевтично прийнятна сіль.
2. Сполука за п. 1, яка відрізняється тим, що R2 являє собою Н.
3. Сполука за п. 1 або п. 2, яка відрізняється тим, що R3 являє собою Н, a R4 являє собою метил.
4. Сполука за будь-яким з пп. 1-3, яка відрізняється тим, що X являє собою:
.
5. Сполука за п. 1, яка відрізняється тим, що являє собою:
 ,
,
або її фармацевтично прийнятна сіль.
6. Сполука за п. 5, яка відрізняється тим, що являє собою:
 ,
,
або її фармацевтично прийнятна сіль.
7. Хлористоводнева сіль сполуки за п. 6, яка відрізняється тим, що являє собою:
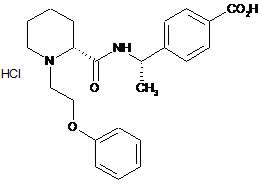 .
.
8. Спосіб лікування остеоартриту в пацієнта, що включає введення пацієнтові, який потребує такого лікування, ефективної кількості сполуки або її фармацевтично прийнятної солі за пп. 1-7.
9. Спосіб лікування ревматоїдного артриту в пацієнта, що включає введення пацієнтові, який потребує такого лікування, ефективної кількості сполуки або її фармацевтично прийнятної солі за пп. 1-7.
10. Спосіб лікування болю, пов'язаного з остеоартритом або ревматоїдним артритом у пацієнта, що включає введення пацієнтові, який потребує такого лікування, ефективної кількості сполуки або її фармацевтично прийнятної солі за пп. 1-7.
11. Сполука або її фармацевтично прийнятна сіль за пп. 1-7 для застосування в терапії.
12. Сполука або її фармацевтично прийнятна сіль за пп. 1-7 для застосування в лікуванні остеоартриту.
13. Сполука або її фармацевтично прийнятна сіль за пп. 1-7 для застосування в лікуванні ревматоїдного артриту.
14. Сполука або її фармацевтично прийнятна сіль за пп. 1-7 для застосування в лікуванні болю, пов'язаного з остеоартритом або ревматоїдним артритом.
15. Фармацевтична композиція, що містить сполуку або її фармацевтично прийнятну сіль за пп. 1-7, у поєднанні з одним або більше фармацевтично прийнятними носіями, розріджувачами або наповнювачами.
16. Фармацевтична композиція за п. 15, яка відрізняється тим, що містить один або більше інших терапевтичних агентів.
Текст
Реферат: У цьому винаході запропонована сполука Формули II: CO2H NH N R O 2 O R X де X являє собою: 3 R 4 ,Формула II UA 114325 C2 (12) UA 114325 C2 H3 C 1 , R 1 2 або , 3 4 R являє собою Н, -CN або F; R являє собою Н або метил; R являє собою Н; та R являє 3 4 собою Н, метил або етил; або R та R , сполучені разом, утворюють циклопропілове кільце; або її фармацевтично прийнятна сіль. UA 114325 C2 5 10 15 20 25 30 Цей винахід має відношення до нових феноксіетилпіперидинових сполук, фармацевтичних композицій, що містять такі сполуки, способів застосування цих сполук для лікування фізіологічних розладів і до проміжних хімічних сполук та способів, придатних для синтезу цих сполук. Цей винахід належить до галузі лікування запальних станів, таких як артрит, у тому числі остеоартрит і ревматоїдний артрит, включаючи біль, пов'язаний з цими станами. Артритом вражено мільйони пацієнтів лише в Сполучених Штатах, і він є однією з основних причин інвалідності. Лікування часто включає застосування NSAID (нестероїдні протизапальні лікарські препарати) або інгібіторів COX-2 (циклооксигеназа-2), які можуть бути джерелом небажаних побічних ефектів з боку серцево-судинної та/або шлунково-кишкової системи. У зв'язку з цим пацієнтам, які мають проблеми, пов'язані з серцево-судинною системою, такі як гіпертонія, може бути протипоказаним застосування NSAID або інгібіторів COX-2. Отже, існує необхідність в альтернативних лікарських препаратах для лікування остеоартриту і ревматоїдного артриту, переважно без побічних ефектів, спричинюваних існуючими на цей час лікарськими препаратами. Відомі чотири підтипи рецепторів простагландину Е2 (PGE2): EP1, EP2, EP3 та EP4. Відомо, що EP4 є основним рецептором, залученим до запального болю суглобів в моделях ревматоїдного артриту і остеоартриту у гризунів (дивись, наприклад, J. Pharmacol. Exp. Ther., 325, 425 (2008)). Отже, селективний антагоніст EP4 може бути корисним в лікуванні артриту, у тому числі артритного болю. Крім того, вважають, що, оскільки антагонізм по відношенню до EP4 не заважає біосинтезу простаноїдів, таких як PGI 2 та ТхА2, селективний антагоніст EP4 може бути нездатним спричинювати серцево-судинні побічні ефекти, що спостерігаються у разі NSAID та інгібіторів СОХ-2 (дивись, наприклад, Bioorganic & Medicinal Chemistry Letters, 21, 484 (2011)). У WO 2013/004290 описані циклічні похідні амінів як антагоністи рецептора EP4. У US 2005/0250818 описані певні ортозаміщені сполуки ариламідів і гетероариламідів, які є селективними антагоністами рецептора EP4 з аналгетичною активністю. Крім того, у WO 2011/102149 описані певні сполуки, які є селективними антагоністами EP4, придатними для лікування IL-23-опосередкованих захворювань. У цьому винаході запропоновані нові сполуки, які є селективними інгібіторами EP4 у зіставленні з EP1, EP2 та EP3. Крім того, у цьому винаході запропоновані нові сполуки з потенціалом зниження побічних ефектів з боку серцево-судинної або шлунково-кишкової системи у порівнянні з традиційними NSAID. Отже, у цьому винаході запропонована сполука Формули II: Формула II Formula II , 35 де X являє собою: , , or або ; 1 40 R являє собою H, -CN, або F; 2 R являє собою H або метил; 3 R являє собою H; та 4 R являє собою H, метил або етил; або 3 4 R та R , сполучені разом, утворюють циклопропілове кільце; або її фармацевтично прийнятна сіль. У цьому винаході також запропонована сполука Формули I: 1 UA 114325 C2 Формула I Formula I 5 10 15 20 25 30 35 40 45 або її фармацевтично прийнятна сіль. У цьому винаході також запропонований спосіб лікування артриту в пацієнта, що включає введення пацієнтові, який потребує такого лікування, ефективної кількості сполуки Формули I або Формули II, або фармацевтично прийнятної солі такої сполуки. У цьому винаході також запропонований спосіб лікування остеоартриту в пацієнта, що включає введення пацієнтові, який потребує такого лікування, ефективної кількості сполуки Формули I або Формули II, або фармацевтично прийнятної солі такої сполуки. Крім того, у цьому винаході також запропонований спосіб лікування ревматоїдного артриту в пацієнта, що включає введення пацієнтові, який потребує такого лікування, ефективної кількості сполуки Формули I або Формули II, або фармацевтично прийнятної солі такої сполуки. У цьому винаході також запропонований спосіб лікування болю, пов'язаного з артритом у пацієнта, що включає введення пацієнтові, який потребує такого лікування, ефективної кількості сполуки Формули I або Формули II, або фармацевтично прийнятної солі такої сполуки. У цьому винаході також запропонований спосіб лікування болю, пов'язаного з остеоартритом або ревматоїдним артритом у пацієнта, що включає введення пацієнтові, який потребує такого лікування, ефективної кількості сполуки Формули I або Формули II, або фармацевтично прийнятної солі такої сполуки. Крім того, у цьому винаході запропонована сполука Формули I або Формули II, або фармацевтично прийнятна сіль такої сполуки, для застосування в терапії, зокрема для лікування остеоартриту. Крім того, у цьому винаході запропонована сполука Формули I або Формули II, або фармацевтично прийнятна сіль такої сполуки, для застосування в лікуванні ревматоїдного артриту. У цьому винаході також запропонована сполука або її фармацевтично прийнятна сіль для застосування в лікуванні болю, пов'язаного з остеоартрозом або ревматоїдним артритом. Крім того, у цьому винаході запропоноване застосування сполуки Формули I або Формули II, або фармацевтично прийнятної солі такої сполуки, для виробництва лікарського засобу для лікування остеоартриту. У цьому винаході запропоноване застосування сполуки Формули I або Формули II, або фармацевтично прийнятної солі такої сполуки, для виробництва лікарського засобу для лікування ревматоїдного артриту. У цьому винаході також запропоноване застосування сполуки Формули I або Формули II, або фармацевтично прийнятної солі такої сполуки, для виробництва лікарського засобу для лікування болю, пов'язаного з остеоартрозом або ревматоїдним артритом. У цьому винаході також запропонована фармацевтична композиція, яка містить сполуку Формули I або Формули II, або фармацевтично прийнятну сіль такої сполуки, у поєднанні з одним або більше фармацевтично прийнятним(-ми) носієм(-ями), розріджувачем(-ами) або наповнювачем(-ами). У конкретному варіанті здійснення цього винаходу згадана композиція також містить один або більше інший(-их) лікарський(-их) засіб(-обів). У цьому винаході також охоплені нові проміжні продукти та способи синтезу сполуки Формули I або Формули II, або фармацевтично прийнятної солі такої сполуки. Крім того, у цей винахід включений спосіб лікування запальних станів таких як артрит, у тому числі остеоартроз та ревматоїдний артрит, у пацієнта, який включає введення пацієнтові, який потребує такого лікування, ефективної кількості антагоніста простагландину, який спричинює запальний стан, такого як антагоніст EP4, у поєднанні з ефективною кількістю модулятора ліпоксину або резолвінового рецептора, такого як модулятор BLT-1, BLT-2, ALX/FPR1, GPR32, CysLT1, CysLT2 або ChemR23. Один із аспектів цього винаходу включає спосіб лікування запального захворювання такого як артрит, у тому числі остеоартрозу та ревматоїдного артриту, у пацієнта, який включає введення пацієнтові, який потребує такого лікування, ефективної кількості інгібітору сінтази простагландину, який спричинює запальний стан, такого як інгібітор mPGES-1, у поєднанні з 2 UA 114325 C2 5 10 15 20 25 30 35 40 ефективною кількістю модулятора ліпоксину або резолвінового рецептора, такого як модулятор BLT-1, BLT-2, ALX/FPR1, GPR32, CysLT1, CysLT2 або ChemR23. Термін "лікування" або "лікувати", вжитий у цьому описі, означає перешкоджання, стримування, уповільнення, зупинення або звернення розвитку або тяжкості існуючого симптому або розладу. Термін "пацієнт", вжитий у цьому описі, означає ссавця, такого як миша, морська свинка, пацюк, собака або людина. Зрозуміло, що переважно пацієнтом є людина. Термін "ефективна кількість", вжитий у цьому описі, означає кількість або дозу сполуки за цим винаходом, або її фармацевтично прийнятної солі, що при введенні згаданому пацієнтові разовою дозою або множинною дозою забезпечує бажаний вплив на пацієнта при призначенні лікування або при лікуванні. Ефективна кількість може бути легко визначена лікарем, який призначає лікування, як фахівцем у цій галузі, із застосуванням відомих методик і внаслідок спостереження результатів, одержаних за аналогічних обставин. При визначенні ефективної для пацієнта кількості лікар, який призначає лікування, розглядає низку факторів, у тому числі, але без обмеження ними: вид ссавця; розмір, вік і загальний стан здоров'я особини; конкретне захворювання або пов'язаний з ним розлад; ступінь або ураження, або тяжкість захворювання або розладу; індивідуальну реакцію пацієнта; конкретну призначену сполуку; спосіб введення; характеристики біодоступності введеного засобу; вибрану схему прийому лікарського засобу; застосування супутніх лікарських засобів; та інші важливі обставини. Сполуки Формули I або Формули II, або їх фармацевтично прийнятна сіль, зазвичай є ефективними в широкому діапазоні доз. Наприклад, добові дози зазвичай становлять від приблизно 0,01 мг на кг маси тіла до приблизно 50 мг на кг маси тіла. У деяких випадках дози нижче нижньої межі згаданого діапазону можуть бути більш ніж достатніми, тоді як в інших випадках можуть бути використані ще більш високі дози без спричинення жодних шкідливих побічних ефектів, і, отже, згаданий діапазон доз жодним чином не є призначеним для обмеження обсягу цього винаходу. Сполуки за цим винаходом виготовляють переважно у вигляді фармацевтичних композицій, які вводять будь-яким способом, який робить згадану сполуку біодоступною. За варіантом, якому віддають найбільшу перевагу, такі композиції призначені для перорального введення. Такі фармацевтичні композиції та способи їх одержання є добре відомими в цій галузі (дивись, наприклад, Remington: The Science and Practice of Pharmacy (Troy, D.B., Editor, 21st Edition, Lippincott, Williams & Wilkins, 2006)). Сполуки Формули I та Формули II є дуже корисними в способах лікування за цим винаходом, при цьому для сполук Формули I та Формули II перевагу віддають певним групам, замісникам та структурам. Такі групи, замісники та структури, яким віддають перевагу, наведені нижче. Зрозуміло, що ці групи, замісники та структури, яким віддають перевагу, є придатними для застосування як у способах лікування, так і в нових сполуках за цим винаходом. 1 2 Перевагу віддають варіанту здійснення цього винаходу, у якому R являє собою H, R являє 3 собою H, R являє собою H, та X являє собою . 3 Перевагу віддають також варіанту здійснення цього винаходу, у якому, R являє собою H, а 4 R являє собою метил. Перевагу віддають також варіанту здійснення цього винаходу, у якому X являє собою 45 . Особливу перевагу віддають 4-[(1S)-1-[[(2R)-1-(2-феноксіетил)піперидин-2карбоніл]аміно]етил]бензойній кислоті наведеної нижче структури: 3 UA 114325 C2 5 10 15 20 25 30 35 40 45 та її фармацевтично прийнятній солі. Особливу перевагу віддають також гідрохлориду 4-[(1S)-1-[[(2R)-1-(2-феноксіетил)піперидин2-карбоніл]аміно]етил]бензойної кислоти. Термін "kPag", вжитий у цьому описі, означає надлишковий тиск у кПа; "Boc" означає третбутоксикарбонілову захисну групу; "DMEM" означає модифіковане за способом Дульбекко середовище Ігла; "ACN" означає ацетонітрил; "DMSO" означає диметилсульфоксид; "DMF" означає N,N-диметилформамід; "EtOH" означає етанол; "THF" означає тетрагідрофуран; "MeOH" означає метанол; "EtOAc" означає етилацетат; "Et2O" означає діетиловий ефір; "TBME" означає трет-бутилметиловий простий ефір; "BOP" означає бензотріазол-1ілокситрис(диметиламіно)фосфонію гексафторофосфат; "NaHMDS" означає натрію біс(триметилсиліл)амід; "PGE2" означає простагландин E2; "FBS" означає ембріональну бичачу сироватку (Fetal Bovine Serum); "IBMX" означає (3-ізобутил-1-метилксантин); "MES" означає (2(N-морфоліно)етансульфонова кислота; "HEPES" означає (2-[4-(2-гідроксіетил)піперазин-1іл]етансульфонова кислота); "HTRF" означає метод роздільно-часової флуоресценції гомогенного середовища; "HEK" означає культуру клітин нирки людського ембріону; "HBSS" означає сбалансований сольовий розчин Хенка; "EC80" означає концентрацію речовини, яка спричинює 80 % максимальної дієвості, можливої для цієї речовини; та "IC50" означає концентрацію засобу, яка спричинює 50 % максимальної інгібувальної реакції, можливої для цієї речовини. Фармацевтично прийнятні солі та традиційні методи їх одержання добре відомі в цій галузі. Дивись, наприклад, Gould, P.L., "Salt selection for basic drugs", International Journal of Pharmaceutics, 33: 201-217 (1986); Bastin, R.J., et al. "Salt Selection and Optimization Procedures for Pharmaceutical New Chemical Entities", Organic Process Research and Development, 4: 427-435 (2000) та Berge, S.M., et al. "Pharmaceutical Salts", Journal of Pharmaceutical Sciences, 66: 1-19, (1977). Фахівцеві з синтезу зрозуміло, що сполуки Формули I та Формули II можна легко перетворити на певну фармацевтично прийнятну сіль та виділити у вигляді цієї фармацевтично прийнятної солі, такої як гідрохлорид, із застосуванням технологічних прийомів та умов, добре відомих фахівцеві в цій галузі. Крім того, фахівцеві з синтезу зрозуміло, що сполуки Формули I та Формули II можна легко перетворити на відповідну вільну основу або вільну кислоту та виділити у вигляді цієї вільної основи або вільної кислоти з відповідної фармацевтично прийнятної солі. У цьому винаході охоплені всі окремі енантіомери або діастереомери, а також суміші енантіомерів і діастереомерів згаданих сполук, у тому числі рацемати. Окремі ізомери, енантіомери або діастереомери можуть бути відокремлені або розділені фахівцем у цій галузі в будь-якій зручній точці синтезу сполук за цим винаходом такими методами, як селективна кристалізація або хіральна хроматографія (дивись, наприклад, Jacques, J. et al., "Еnantiomers, Racemates, and Resolutions", John Wiley and Sons, Inc., 1981 та Eliel, E.L. and Wilеn, S.H., "Stereochemistry of Organic Compounds", Wiley-Intersciеnce, 1994). Сполуку за цим винаходом або її фармацевтично прийнятні солі можна одержати в різні способи, відомі в цій галузі, деякі з яких показані в наведених нижче Схемах, Препаративних методиках та Прикладах. Для одержання сполуки Формули I або її фармацевтично прийнятної солі можна комбінувати різноманітними способами конкретні стадії синтезу кожного із описаних шляхів або їх можна здійснюватись у поєднанні зі стадіями різних схем. Продукти кожної стадії наведених нижче схем можуть бути виділені традиційними методами, у тому числі, екстрагуванням, випарюванням, осадженням, хроматографуванням, фільтруванням, розтиранням в порошок та кристалізацією. Реагенти та вихідні матеріали є легко доступними для фахівця в цій галузі. Усі замісники, якщо не зазначено інше, відповідають визначенням, 4 UA 114325 C2 наведеним вище. Зрозуміло, що ці Схеми, Препаративні методики та Приклади жодним чином не призначені для обмеження обсягу цього винаходу. Схема 1 HCl Стадія А Step A 5 10 15 20 Стадія Step B В Стадія Step C С Препаративна методика 1 Синтез (S)-N-трет-бутоксикарбоніл-1-(4-бромофеніл)етиламіну Схема 1, Стадія A: до перемішуваного розчину (-)-1-(4-бромофеніл)етиламіну (1,00 г, 5,0 ммоль) у дихлорометані (10 мл) при 0 °C додають ди-трет-бутилдикарбонат (1,09 г, 5,0 ммоль). Цю суміш лишають для нагрівання до кімнатної температури, потім перемішують протягом двох годин. До згаданої перемішуваної суміші додають 1 M водний розчин соляної кислоти (25 мл), а потім Et2O (25 мл). Шари розділяють, та водний шар екстрагують Et 2O (225 мл). Об'єднують органічні шари, промивають насиченим водним розчином NaCl (25 мл), сушать органічні шари над MgSO4, фільтрують для видалення твердих речовин, концентрують фільтрат під зниженим тиском, і одержують вказану в заголовку сполуку у вигляді твердої речовини білого кольору 79 81 + + 1 (1,50 г, вихід 99 %). Масс-спектр (m/z) ( Br/ Br) 244/246 (M+2H-t-Bu) , 322/324 (M+Na) . H ЯМР (400 МГц, CDCl3): δ 7,46-7,43 (m, 2H), 7,19-7,15 (m, 2H), 4,81-4,65 (m, 1H), 1,48-1,36 (m, 12H). Наведену нижче сполуку одержують по суті за способом, описаним у Препаративній методиці 1, із використанням 1-(4-бромофеніл)циклопропанаміну замість (-)-1-(4бромофеніл)етиламіну: Препар. методика Хімічна назва Структура MS (m/z) 2 79 трет-бутил-N-[1-(4бромофеніл) циклопропіл]карбамат 81 ( Br/ Br) 256/258 + (M+2H-t-Bu) , + 334/336 (M+Na) Препаративна методика 3 Синтез метил-(S)-4-(1-трет-бутоксикарбоніламіноетил)бензоату 25 30 35 40 Схема 1, Стадія B: Pd(OAc)2 (120 мг, 0,53 ммоль), 1,1'-біс(дифенілфосфіно)ферроцен (355 мг, 0,64 ммоль), (S)-N-трет-бутоксикарбоніл-1-(4-бромофеніл)етиламін (1,50 г, 5,0 ммоль), безводний CH3CN (45 мл), безводний CH3OH (30 мл) та триетиламін (1,9 мл, 13,63 ммоль) додають у автоклав Парра з механічною мішалкою. Згадану посудину герметизують, та моноксидом вуглецю підвищують тиск до досягнення надлишкового тиску 724 кПа. Згадану посудину нагрівають до 85 °C, та перемішують суміш протягом ночі. Відкривають реакційну посудину (Увага – отруйний газ!), та її вміст переносять до круглодонної колби із промиванням CH3OH. Суміш концентрують під зниженим тиском до одержання залишку помаранчевого кольору. Додають воду (50 мл), потім екстрагують EtOAc (250 мл). Об'єднані органічні фази промивають насиченим водним розчином NaCl (25 мл), потім шари розділяють, згадану органічну фазу сушать над MgSO4, фільтрують для видалення твердих речовин, фільтрат концентрують під зниженим тиском, і одержують неочищений продукт. Цей продукт очищують засобами флеш-хроматографії на силікагелі, елююючи з градієнтом від 0 % до 60 % розчину EtOAc/гексани. Фракції, які містять бажаний продукт, концентрують під зниженим тиском, і одержують вказану в заголовку сполуку у вигляді твердої речовини білого кольору (1,00 г, 72 % + + + 1 вихід). Масс-спектр (m/z) 224 (M+2H-t-Bu) , 302 (M+Na) , 581 (2M+Na) . H ЯМР (400 МГц, DMSO-d6): δ 7,89 (d, J=8,4 Гц, 2H), 7,41 (d, J=8,2 Гц, 2H), 4,64 (dq, J=7,4 Гц, 6,8 Гц, 1H), 3,82 (s, 5 UA 114325 C2 3H), 1,34 (широкий s, 9H), 1,28 (d, J=7,2 Гц, 3H). Наведену нижче сполуку одержують по суті за способом, описаним у Препаративній методиці 3, із використанням трет-бутил-N-[1-(4-бромофеніл)циклопропіл]карбамату замість (S)N-трет-бутоксикарбоніл-1-(4-бромофеніл)етиламіну: 5 Препар. методика Хімічна назва Структура 4 метил-4-[1-(третбутоксикарбоніламіно)циклопропіл]бензоат MS (m/z) + 236 (M+2H-t-Bu) , + 314 (M+Na) , 605 + (2M+Na) Препаративна методика 5 Синтез гідрохлориду метил-(S)-4-(1-аміноетил)бензоату HCl 10 15 Схема 1, Стадія C: до метил-(S)-4-(1-трет-бутоксикарбоніламіноетил)бензоату (1,00 г, 3,58 ммоль) додають хлористий водень (4 M розчин у диоксані, 5 мл, 20 ммоль), та перемішують одержану суміш при кімнатній температурі протягом однієї години. Цю суміш концентрують під зниженим тиском, і одержують вказану в заголовку сполуку у вигляді твердої речовини білого 1 кольору (750 мг, вихід 97 %). H ЯМР (400 МГц, DMSO-d6): (8,57 (широкий s, 3H), 7,99 (d, J=8,4 Гц, 2H), 7,65 (d, J=8,4 Гц, 2H), 4,47 (q, J=6,7 Гц, 1H), 3,84 (s, 3H), 1,50 (d, J=6,8 Гц, 3H). Наведену нижче сполуку одержують по суті за способом, описаним в Препаративній методиці 5, із використанням метил-4-[1-(трет-бутоксикарбоніламіно)циклопропіл]бензоату замість метил-(S)-4-(1-трет-бутоксикарбоніламіноетил)бензоату: Препар. методика Хімічна назва Структура 6 гідрохлорид метил-4-(1аміноциклопропіл)бензоату HCl MS (m/z) 192 (M+H) + 20 Схема 2 Step A А Стадія Препаративна методика 7 Синтез 1-(2,2-диметоксіетокси)-4-фторобензолу 25 30 Схема 2, Стадія A: 4-фторофенол (5,5 г, 49,1 ммоль) розчиняють у ацетонітрилі (49 мл), цей розчин обробляють 2-бромо-1,1-диметоксіетаном (11,6 мл, 98,1 ммоль) та K2CO3 (16,95 г, 122,7 ммоль). Розчин нагрівають зі зворотним холодильником при перемішуванні протягом п'яти діб. Цю суміш фільтрують, та одержаний фільтрат концентрують під зниженим тиском. Одержаний неочищений матеріал очищують засобами хроматографії на силікагелі, елююючи з градієнтом від 0 % до 50 % розчину EtOAc/гексани. Фракції, які містять бажаний продукт, концентрують під 6 UA 114325 C2 зниженим тиском, і одержують вказану в заголовку сполуку у вигляді безбарвного масла (5,85 г, + + вихід 60 %). Масс-спектр (m/z) 218 (M+NH4) , 223 (M+Na) . Схема 3 Step A А Стадія 5 10 15 20 Препаративна методика 8 Синтез (2,2-диметоксіетокси)циклогексану Схема 3, Стадія A: циклогексанол (2,00 мл, 19,1 ммоль) розчиняють у DMF (9,6 мл), потім додають NaHMDS (1 M розчин у THF, 21,0 мл, 21,0 ммоль), і перемішують цей розчин при кімнатній температурі протягом 5 хв. Додають 2-бромо-1,1-диметоксіетан (2,26 мл, 19,1 ммоль), потім цю суміш перемішують при кімнатній температурі в атмосфері азоту протягом трьох діб. Цю суміш розбавляють EtOAc (250 мл), і промивають насиченим водним розчином NaCl (2250 мл). Органічну фазу сушать над MgSO4, фільтрують, та одержаний фільтрат концентрують під зниженим тиском. Одержаний неочищений матеріал очищають засобами хроматографії на силікагелі, елююючи з градієнтом від 0 % до 10 % розчину EtOAc/гексани. Фракції, які містять бажаний продукт, концентрують під зниженим тиском, і одержують вказану в заголовку сполуку 1 у вигляді блідо-жовтого масла (1,10 г, вихід 31 %). H ЯМР (400 МГц, CDCl3): δ 4,47 (t, J=5,3 Гц, 1H), 3,49 (d, J=5,3 Гц, 2H), 3,39 (s, 6H), 3,25 (tt, J=9,2 Гц, 3,7 Гц, 1H), 1,94-1,87 (m, 2H), 1,76-1,69 (m, 2H), 1,55-1,49 (m, 1H), 1,34-1,17 (m, 5H). Схема 4 Стадія Step A А Препаративна методика 9 Синтез 2-(4-фторофенокси)ацетальдегіду 25 30 Схема 4, Стадія A: 1-(2,2-диметоксіетокси)-4-фторобензол (1,00 г, 4,99 ммоль) розчиняють у хлороформі (5,0 мл), та цю суміш обробляють трифторооцтовою кислотою (0,755 мл, 9,99 ммоль). Одержану суміш перемішують при кімнатній температурі протягом двох діб, потім нагрівають до 65 °C, і перемішують протягом 4 год. Цю суміш концентрують під зниженим тиском, і одержують вказану в заголовку сполуку у вигляді безбарвного масла з чистотою 1 1 приблизно 70 % за результатами H ЯМР дослідження (550 мг, 71 % невідкоригований вихід). H ЯМР (400 МГц, CDCl3): δ 9,85 (t, J=0,5 Гц, 1H), 7,00 (dd, J=8,9 Гц, 8,5 Гц, 2H), 6,85 (dd, J=9,5 Гц, 4,3 Гц, 2H), 4,55 (широкий s, 2H). Схема 5 Стадія Step A А 7 UA 114325 C2 Препаративна методика 10 Синтез 2-(циклогексилокси)ацетальдегіду 5 10 Схема 5, Стадія A: суміш (2,2-диметоксіетокси)циклогексану та води (30 мл) підкислюють до pH 1,0 сірчаною кислотою (9,0 M водний розчин), та подають цю суміш на головку для молекулярної перегонки. Тиск зменшують до 26,7 кПа, і суміш нагрівають до 100 °C протягом 1 год. Суміш охолоджують до кімнатної температури, потім водний шар екстрагують TBME (275 мл). Об'єднані органічні шари промивають насиченим водним розчином NaHCO 3 (75 мл) та насиченим водним розчином NaCl (75 мл). Органічну фазу сушать над MgSO 4, фільтрують, фільтрат концентрують під зниженим тиском, і одержують вказану в заголовку сполуку (634 мг, 1 вихід 51 %). H ЯМР (400 МГц, CDCl3): δ 9,73 (t, J=1,0 Гц, 1H), 4,06 (d, J=1,0 Гц, 2H), 3,31 (tt, J=9,2 Гц, 3,9 Гц, 1H), 1,95-1,89 (m, 2H), 1,79-1,68 (m, 2H), 1,57-1,50 (m, 1H), 1,39-1,20 (m, 5H). Схема 6 Step A Стадія 15 20 25 30 А Step B Стадія В HCl Препаративна методика 11 Синтез метил-(R)-1-(2-феноксіетил)піперидин-2-карбоксилату Схема 6, Стадія A: метил-(R)-піперидин-2-карбоксилат (5,00 г, 34,9 ммоль) розчиняють у DMF (87 мл), і обробляють K2CO3 (14,48 г, 104,8 ммоль) та β-бромофенетолом (7,16 г, 34,9 ммоль). Одержану суміш перемішують протягом ночі при 100 °C. Цю суміш охолоджують до кімнатної температури, і додають EtOAc (250 мл). Органічну фазу промивають водою (4100 мл) і насиченим водним розчином NaCl (100 мл). Органічну фазу сушать над K 2CO3, фільтрують для видалення твердих речовин, одержаний фільтрат концентрують під зниженим тиском, і одержують масло жовтого кольору. Цей неочищений матеріал очищують засобами флешхроматографії на силікагелі, елююючи з градієнтом від 20 % до 100 % розчину EtOAc/гексани. Фракції, які містять бажаний продукт, концентрують під зниженим тиском, і одержують вказану в 1 заголовку сполуку у вигляді безбарвного масла з чистотою приблизно 90 % за результатами H + 1 ЯМР дослідження (6,50 г, вихід 64 %). Масс-спектр (m/z) 264 (M+H) . H ЯМР (400 МГц, CDCl3): δ 7,27 (dd, J=8,5 Гц, 7,3 Гц, 2H), 6,93 (t, J=7,3 Гц, 1H), 6,88 (d, J=8,8 Гц, 2H), 4,10 (app t, J=6,1 Гц, 2H), 3,72 (s, 3H), 3,29 (dd, J=8,4 Гц, 4,0 Гц, 1H), 3,14 (app dt, J=11,3 Гц, 6,4 Гц, 1H), 2,95 (app dt, J=13,7 Гц, 6,1 Гц, 1H), 2,89 (app dt, J=13,7 Гц, 6,1 Гц, 1H), 2,42 (ddd, J=11,6 Гц, 7,9 Гц, 3,7 Гц, 1H), 1,90-1,82 (m, 1H), 1,79 (app td, J=8,9 Гц, 3,8 Гц, 1H), 1,67-1,59 (m, 3H), 1,39 (app td, J=8,8 Гц, 4,0 Гц, 1H). 8 UA 114325 C2 Препаративна методика 12 Синтез гідрохлориду (R)-1-(2-феноксіетил)піперидин карбонової кислоти HCl 5 10 15 Схема 6, Стадія B: метил-(R)-1-(2-феноксіетил)піперидин-2-карбоксилат (6,50 г, 22,2 ммоль) розчиняють у THF (11,1 мл) при кімнатній температурі, додають NaOH (5 M водний розчин, 8,89 мл, 44,4 ммоль), і нагрівають при перемішуванні до 65 °C протягом ночі. Додають хлористий водень (5 M водний розчин), доки pH згаданої водної фази не досягне 1,0. Цю водну фазу промивають CH2Cl2 (375 мл). Згадану водну фазу концентрують під зниженим тиском до одержання твердої речовини білого кольору. Цю тверду речовину розтирають з EtOH (50 мл), фільтрують для видалення суспедованих солей, фільтрат концентрують під зниженим тиском, і одержують вказану в заголовку сполуку у вигляді твердої речовини білого кольору (5,17 г, вихід + 1 81 %). Масс-спектр (m/z) 250 (M+H) . H ЯМР (400 МГц, CD3OD): δ 7,31 (dd, J=8,8 Гц, 7,5 Гц, 2H), 7,02-6,97 (m, 3H), 4,47 (AB-зв'язані ddd, J=11,5 Гц, 7,1 Гц, 3,0 Гц, 1H), 4,38 (AB-зв'язані ddd, J=11,8 Гц, 6,3 Гц, 3,1 Гц, 1H), 4,15 (dd, J=11,2 Гц, 2,8 Гц, 1H), 3,80 (d, J=12,2 Гц, 1H), 3,73-3,68 (m, 2H), 3,29 (app td, J=13,2 Гц, 3,8 Гц, 1H), 2,35 (d, J=13,5 Гц, 1H), 2,00-1,80 (m, 4H), 1,68 (app t, J=13,1 Гц, 1H). Схема 7 HCl 20 25 30 35 Стадія Step A HCl А Препаративна методика 13 Синтез метил-4-[(1S)-1-[[(2R)-1-(2-феноксіетил)піперидин-2-карбоніл]аміно]етил]бензоату Схема 7, Стадія A: гідрохлорид (R)-1-(2-феноксіетил)піперидин карбонової кислоти (750 мг, 2,62 ммоль) та гідрохлорид метил-(S)-4-(1-аміноетил)бензоату (566 мг, 2,62 ммоль) розчиняють у DMF (5,25 мл) при кімнатній температурі. Додають триетиламін (1,65 мл, 11,81 ммоль), потім BOP (1,51 г, 3,41 ммоль). Цю суміш перемішують при кімнатній температурі протягом 3 год., потім розбавляють EtOAc (25 мл). Промивають суміш насиченим водним розчином LiCl (225 мл). Органічні шари сушать над MgSO4, фільтрують для видалення твердих речовин, та концентрують під зниженим тиском. Одержане масло жовто-помаранчевого кольору очищують засобами флеш-хроматографії на силікагелі, елююючи з градієнтом від 0 % до 100 % розчину EtOAc/гексани. Фракції, які містять бажаний продукт, концентрують під зниженим тиском, і одержують вказану в заголовку сполуку у вигляді твердої речовини білого кольору (930 мг, вихід + 1 86 %). Масс-спектр (m/z) 411 (M+H) . H ЯМР (400 МГц, DMSO-d6): δ 8,13 (d, J=8,0 Гц, 1H), 7,86 (d, J=8,3 Гц, 2H), 7,43 (d, J=8,3 Гц, 2H), 7,24 (dd, J=8,8 Гц, 7,4 Гц, 2H), 6,89 (t, J=8,3 Гц, 1H), 6,85 (dd, J=8,8 Гц, 1,0 Гц, 2H), 4,96 (app пентет, J=7,2 Гц, 1H), 4,05-3,98 (m, 2H), 3,82 (s, 3H), 3,11 (app dt, J=11,4 Гц, 3,7 Гц, 1H), 2,81 (dd, J=7,2 Гц, 2,8 Гц, 1H), 2,77 (app q, J=6,8 Гц, 1H), 2,50 (app dt, J=11,2 Гц, 6,8 Гц, 1H), 2,15 (app td, J=11,6 Гц, 2,8 Гц, 1H), 1,68-1,61 (m, 2H), 1,59-1,40 (m, 3H), 9 UA 114325 C2 1,36 (d, J=7,0 Гц, 3H), 1,27-1,18 (m, 1H). Наведені нижче сполуки одержують по суті за способом, описаним в Препаративній методиці 13, із використанням придатних амонієвих солей замість гідрохлориду метил-(S)-4-(1аміноетил)бензоату: 5 Препар. методика 14 Хімічна назва Структура MS (m/z) 397 + (M+H) метил-4-[[[(2R)-1-(2феноксіетил)піперидин-2карбоніл]аміно]метил]бензоат 15 метил-4-[(1S)-1-[[(2R)-1-(2феноксіетил)піперидин-2карбоніл]аміно]пропіл]бензоат 425 + (M+H) 16 метил-4-[1-[[(2R)-1-(2феноксіетил)піперидин-2карбоніл]аміно]циклопропіл]бензоат 423 + (M+H) Схема 8 Стадія А Step A Стадія Step B В Стадія Step C С 10 UA 114325 C2 Препаративна методика 17 Синтез карбоніл]аміно]етил]бензоату 5 10 15 20 25 30 35 40 метил-4-[(1S)-1-[[(2R)-піперидин-1-трет-бутоксикарбоніл-2 Схема 8, Стадія A: до суміші (R)-N-трет-бутоксикарбонілпіпеколінової кислоти (20,0 г, 87,2 ммоль) та CH2Cl2 (400 мл), температура якої становить 0 °C, додають триетиламін (13,4 мл, 96,0 ммоль). Потім краплинами додають ізобутилхлороформіат (12,5 мл, 96,0 ммоль), та перемішують протягом 20 хв. Додають метил-4-[(S)-аміноетил]бензоат (17,2 г, 96,0 ммоль), потім одержану суміш залишають для нагрівання до кімнатної температури, та перемішують протягом однієї години. Додають воду (300 мл), потім шари розділяють, органічний шар промивають 1 M водним розчином KHSO4 (200 мл), потім насиченим водним розчином NaCl (200 мл). Органічний шар відокремлюють, та сушать над MgSO 4, потім фільтрують для видалення твердих речовин, фільтрат концентрують під зниженим тиском, і одержують вказану в заголовку сполуку у вигляді безбарвного масла (34,0 г, вихід 100 %). Масс-спектр (m/z) 291 (M+ + 1 Boc+2H) , 413 (M+Na) . H ЯМР (300 МГц, CDCl3): δ 7,99 (d, J=8,2 Гц, 2H), 7,33 (d, J=8,2 Гц, 2H), 5,14 (app пентет, J=7,1 Гц, 1H), 4,72 (широкий s, 1H), 3,90 (s, 3H), 3,40 (dd, J=6,2 Гц, 5,8 Гц, 1H), 2,63 (app td, J=6,9 Гц, 2,4 Гц, 1H), 2,25 (широкий s, 1H), 1,75 (app tq, J=13,4 Гц, 6,7 Гц, 1H), 1,631,50 (m, 3H), 1,47 (s, 9H), 1,38 (app t, J=5,6 Гц, 1H), 0,91 (d, J=6,6 Гц, 3H). Препаративна методика 18 Синтез метил-4-[(1S)-1-[[(2R)-піперидин-2-карбоніл]аміно]етил]бензоату Схема 8, Стадія B: до суміші EtOAc (136 мл) та EtOH (55,8 мл), температура якої становить 0 °C, краплинами додають ацетилхлорид (62,0 мл, 871 ммоль), потім цю суміш лишають для нагрівання до кімнатної температури протягом 30 хв, додають розчин метил-4-[(1S)-1-[[(2R)піперидин-1-трет-бутоксикарбоніл-2-карбоніл]аміно]етил]бензоату (34,0 г, 87,1 ммоль) у EtOAc (136 мл), потім перемішують цю реакційну суміш при кімнатній температурі протягом однієї години. Екстрагують суміш водою (2100 мл), потім до об'єднаних водних шарів додають 32 % водний розчин аміаку, доки pH не досягне 10. Цю суміш екстрагують TBME (2200 мл), потім об'єднані органічні шари сушать над MgSO 4, фільтрують для видалення твердих речовин, фільтрат концентрують під зниженим тиском, і одержують вказану в заголовку сполуку у вигляді + + твердої речовини білого кольору (20,2 г, вихід 80 %). Масс-спектр (m/z) 291 (M+H) , 581 (2M+H) . 1 H ЯМР (300 МГц, CDCl3): δ 8,00 (d, J=8,5 Гц, 2H), 7,37 (d, J=8,2 Гц, 2H), 7,15 (широкий d, J=7,6 Гц, 1H), 5,14 (app пентет, J=7,4 Гц, 1H), 3,90 (s, 3H), 3,23 (dd, J=9,9 Гц, 3,3 Гц, 1H), 3,01 (app dt, J=11,8 Гц, 3,5 Гц, 1H), 2,68 (ddd, J=12,1 Гц, 10,7 Гц, 3,0 Гц, 1H), 1,98-1,90 (m, 1H), 1,61-1,52 (m, 1H), 1,48 (d, J=7,1 Гц, 3H), 1,43-1,34 (m, 2H). Препаративна методика 19 Синтез метил-4-[(1S)-1-[[(2R)-1-(2-феноксіетил)піперидин-2-карбоніл]аміно]етил]бензоату Схема 8, Стадія C: до суспензії силікагелю (100 г) у CH 2Cl2 (705 мл) при кімнатній температурі додають краплинами розчин NaIO4 (35,0 г, 161,9 ммоль) у воді (235 мл). Цю суміш перемішують протягом 30 хв, потім додають 1,2-дигідрокси-3-феноксипропан (21,5 г, 121,4 ммоль), та перемішують протягом ще 30 хв. Одержану суміш фільтрують для видалення 11 UA 114325 C2 5 10 15 твердих речовин, і шари фільтрату розділяють. Органічні шари сушать над MgSO 4, і фільтрують для видалення твердих речовин. До фільтрату додають метил-4-[(1S)-1-[[(2R)-піперидин-2карбоніл]аміно]етил]бензоат (23,5 г, 80,9 ммоль), а потім маленькими порціями триацетоксиборогідрид натрію (35,7 г, 161,9 ммоль), перемішують протягом однієї години при кімнатній температурі, потім додають 32 % водний розчин аміаку, доки pH не досягне 10. Шари розділяють, та сушать органічну фазу над MgSO 4, фільтрують для видалення твердих речовин, потім фільтрат концентрують під зниженим тиском, і одержують неочищений матеріал. Цей матеріал розчиняють у EtOAc (300 мл), і фільтрують через шар силікагелю (30 г). Фільтрат концентрують під зниженим тиском до одержання 36 г матеріалу. Додають TBME (180 мл), та нагрівають до 50 °C. Підтримуючи температуру на рівні 50 °C, протягом 15 хв додають гексани (360 мл), потім суміш перемішують протягом однієї години. Суміш лишають для охолодження до кімнатної температури, потім тверді речовини виділяють фільтруванням, сушать під зниженим тиском, і одержують вказану в заголовку сполуку у вигляді твердої речовини білого кольору (16,7 г, вихід 50 %). Схема 9 Стадія Step A А HCl Стадія В Step B Препаративна методика 20 Синтез гідрохлориду метил-4-[(1S)-1-[[(2R)-піперидин-2-карбоніл]аміно]етил]бензоату HCl 20 25 Схема 9, Стадія A: метил-4-[(1S)-1-[[(2R)-піперидин-1-трет-бутоксикарбоніл-2карбоніл]аміно]етил]бензоат (7,80 г, 19,98 ммоль) обробляють соляною кислотою (4 M розчин у 1,4-діоксані, 25,0 мл, 99,9 ммоль), одержану суміш перемішують при кімнатній температурі протягом 1 год. Одержану суміш концентрують під зниженим тиском, і одержують вказану в заголовку сполуку у вигляді твердої речовини білого кольору (6,0 г, вихід 92 %). Масс-спектр + + + (m/z) 291 (M+H) , 581 (2M+H) , 603 (2M+Na) . 12 UA 114325 C2 Препаративна методика 21 Синтез карбоніл]аміно]етил]бензоату 5 10 15 метил-4-[(1S)-1-[[(2R)-1-(2-(4-фторофенокси)етил)піперидин-2 Схема 9, Стадія B: суміш гідрохлориду метил-4-[(1S)-1-[[(2R)-піперидин-2карбоніл]аміно]етил]бензоату (650 мг, 1,99 ммоль) та 2-(4-фторофенокси)ацетальдегіду (337 мг, 2,19 ммоль) у DCE (9,9 мл) перемішують при кімнатній температурі протягом 30 хв. Додають оцтову кислоту (0,113 мл, 1,99 ммоль) та триацетоксиборогідрид натрію (590 мг, 2,78 ммоль), і перемішують при кімнатній температурі протягом трьох діб. Реакцію зупиняють насиченим водним розчином NaHCO3 (25 мл), і водний шар екстрагують EtOAc (225 мл). Об'єднані органічні шари промивають насиченим водним розчином NaCl (25 мл), потім органічну фазу сушать над MgSO4, фільтрують, і концентрують під зниженим тиском. Одержане масло очищують засобами флеш-хроматографії на силікагелі, елююючи з градієнтом від 0 % до 100 % розчину EtOAc/гексани. Фракції, які містять бажаний продукт, концентрують під зниженим тиском, і одержують вказану в заголовку сполуку у вигляді твердої речовини білого кольору (600 + + мг, вихід 70 %). Масс-спектр (m/z) 429 (M+H) , 451 (M+Na) . Наведені нижче сполуки одержують по суті за способом, описаним в Препаративній методиці 21, із використанням придатних альдегідів замість 2-(4-фторофенокси)ацетальдегіду: Препар. методика Хімічна назва Структура 22 метил-4-[(1S)-1-[[(2R)-1-(2-(4ціанофенокси)етил)піперидин-2карбоніл]аміно]етил]бензоат MS (m/z) 436 + (M+H) 23 метил-4-[(1S)-1-[[(2R)-1-(2-(2метилфенокси)етил)піперидин-2карбоніл]аміно]етил]бензоат 425 + (M+H) , 447 + (M+Na) 24 метил-4-[(1S)-1-[[(2R)-1-(2циклогексилоксіетил) піперидин-2-карбоніл] аміно]етил]бензоат 417 + (M+H) , 439 + (M+Na) 20 13 UA 114325 C2 Схема 10 Стадія Step A А HCl CF3COOH 5 Препаративна методика 25 Синтез трифтороацетату карбоніл]аміно]етил]бензоату метил-4-[(1S)-1-[[(2R)-1-(1-метил-2-феноксіетил)піперидиній-2 CF3COOH 10 15 Схема 10, Стадія A: суміш гідрохлориду метил-4-[(1S)-1-[[(2R)-піперидин-2карбоніл]аміно]етил]бензоату (150 мг, 0,46 ммоль), 1-фенокси-2-пропанону (69 мкл, 0,50 ммоль), DCE (2,3 мл), оцтової кислоти (26 мкл, 0,46 ммоль) та триацетоксиборогідриду натрію (136 мг, 0,64 ммоль) перемішують при 65 °C протягом двох діб. Реакцію зупиняють насиченим водним розчином NaHCO3 (75 мл), і водний шар екстрагують EtOAc (75 мл). Органічну фазу сушать над Na2SO4, фільтрують, і концентрують під зниженим тиском. Цей неочищений матеріал очищують засобами хроматографії з оберненою фазою на колонці із силікагелем C18, елююючи 0,1 % TFA при градієнті від 5 % до 50 % розчину ACN/вода. Фракції, які містять бажаний продукт, концентрують під зниженим тиском, і одержують вказану в заголовку сполуку у вигляді масла блідо-жовтого кольору як суміші діастереомерів 2:1 (24 мг, вихід 10 %). Масс+ спектр (m/z) 425 (M+H) . Схема 11 Стадія Step A А HCl Приклад 1 Example 1 20 Приклад 1 Синтез гідрохлориду карбоніл]аміно]етил]бензойної кислоти 4-[(1S)-1-[[(2R)-1-(2-феноксіетил)піперидин-2 HCl 25 Схема 11, Стадія A: метил-4-[(1S)-1-[[(2R)-1-(2-феноксіетил)піперидин-2карбоніл]аміно]етил]бензоат (930 мг, 2,27 ммоль) розчиняють у THF (4,0 мл) та CH 3OH (4,0 мл) 14 UA 114325 C2 5 10 15 при кімнатній температурі. Додають NaOH (1 M водний розчин, 4,5 мл, 4,5 ммоль), потім одержану суміш перемішують при кімнатній температурі протягом трьох діб. Цю реакційну суміш концентрують під зниженим тиском до одержання клейкої твердої речовини. Додають хлористий водень (4 M розчин у діоксані, 2 мл, 8 ммоль), та інтенсивно перемішують протягом 10 хв. Суспендовану тверду речовину видаляють фільтруванням, і фільтрат концентрують під зниженим тиском до одержання твердої речовини білого кольору. Цю тверду речовину розтирають у киплячому діетиловому ефірі (25 мл), суспендовану тверду речовину видаляють фільтруванням, і одержують вказану в заголовку сполуку (650 мг, вихід 66 %) у вигляді твердої + 1 речовини білого кольору. Масс-спектр (m/z): 397 (M+H) . H ЯМР (400 МГц, DMSO-d6): δ 12,89 (широкий s, 1H), 10,08 (широкий s, 1H), 9,41 (d, J=7,6 Гц, 1H), 7,88 (d, J=8,2 Гц, 2H), 7,47 (d, J=8,2 Гц, 2H), 7,26 (dd, J=8,4 Гц, 7,6 Гц, 2H), 6,96 (t, J=7,2 Гц, 1H), 6,90 (d, J=7,9 Гц, 2H), 5,02 (app пентет, J=7,1 Гц, 1H), 4,34-4,21 (m, 2H), 4,03 (app t, J=10,2 Гц, 1H), 3,57 (d, J=12,4 Гц, 1H), 3,483,39 (m, 1H), 3,37-3,18 (m, 2H), 2,15 (d, J=13,5 Гц, 1H), 1,82-1,66 (m, 4H), 1,50-1,43 (m, 1H), 1,39 (d, J=7,2 Гц, 3H). Наведені нижче сполуки одержують по суті за способом, описаним у Прикладі 1, із використанням придатних метилових складних ефірів замість метил-4-[(1S)-1-[[(2R)-1-(2феноксіетил)піперидин-2-карбоніл]аміно]етил]бензоату: Приклад Хімічна назва 2 Структура гідрохлорид 4-[[[(2R)-1-(2феноксіетил)піперидин-2карбоніл]аміно]метил]бензойної кислоти MS (m/z) 383 (M+H) + HCl 3 4 5 6 411 (M+H) + 415 (M+H) + 422 (M+H) + HCl гідрохлорид 4-[1-[[(2R)1-(2феноксіетил)піперидин2-карбоніл]аміно]циклопропіл]бензойної кислоти HCl гідрохлорид 4-[(1S)-1[[(2R)-1-(2-(4фторофенокси)етил)піперидин-2-карбоніл]аміно]етил]-бензойної кислоти гідрохлорид 4-[(1S)-1[[(2R)-1-(2-(4ціанофенокси)етил)піперидин-2-карбоніл]аміно]етил]бензойної кислоти + 409 (M+H) гідрохлорид 4-[(1S)-1[[(2R)-1-(2феноксіетил)піперидин2-карбоніл]аміно]пропіл] бензойної кислоти HCl HCl 15 UA 114325 C2 Приклад Хімічна назва 7 гідрохлорид 4-[(1S)-1[[(2R)-1-(2-(2метилфенокси)етил)піперидин-2карбоніл]аміно]етил]бензойної кислоти 8 гідрохлорид 4-[(1S)-1[[(2R)-1-(2циклогексилоксіетил)піперидин-2-карбоніл]аміно]етил]бензойної кислоти Структура MS (m/z) 411 (M+H) + 403 (M+H) + HCl HCl Схема 12 Абсолютний Абсолютний Стадія А Step A CF3COOH 5 CF3COOH Приклад 9 Синтез трифтороацетату 4-[(1S)-1-[[(2R)-1-(1-метил-2-феноксіетил)піперидиній-2карбоніл]аміно]етил]бензойної кислоти Абсолютний CF3COOH 10 15 20 Схема 12, Стадія A: трифтороацетат метил-4-[(1S)-1-[[(2R)-1-(1-метил-2феноксіетил)піперидиній-2-карбоніл]аміно]етил]бензоату (24 мг, 0,045 ммоль) розчиняють у THF (226 мкл), і цю суміш обробляють метанолом (226 мкл) та гідроксидом натрію (1 н. водний розчин, 170 мкл, 0,17 ммоль). Одержану суміш перемішують протягом ночі при кімнатній температурі, потім концентрують під зниженим тиском до одержання клейкої твердої речовини. Цей неочищений матеріал очищують засобами хроматографії з оберненою фазою на колонці з силікагелем C18, елююючи 0,1 % TFA при градієнті від 5 % до 50 % розчину ACN/вода до одержання двох окремих фракцій, кожна з яких містить певний діастереомер продукту. Кожну фракцію концентрують під зниженим тиском, кожну фракцію розчиняють у мінімальному об'ємі метанолу, кожну фракцію розтирають у діетиловому ефірі (5 мл), кожну фракцію концентрують під зниженим тиском, і одержують Ізомер 1 (3,0 мг, вихід 13 %) та Ізомер 2 (1,1 мг, вихід 5 %) вказаної у заголовку сполуки у вигляді твердої речовини білого кольору. + 1 Приклад 9a: основний ізомер (Ізомер 1). Масс-спектр (m/z): 411 (M+H) . H ЯМР (DMSO-d6) δ 9,75 (широкий s), 9,30 (d, J=7,4 Гц, 1H), 7,82 (d, J=7,6 Гц, 2H), 7,43 (d, J=7,6 Гц, 2H), 7,33-7,25 (m, 2H), 7,04-6,95 (m, 3H), 5,03 (app p, J=6,8 Гц, 1H), 4,35-4,24 (m, 2H), 4,07 (dd, J=12,1 Гц, 3,5 Гц, 16 UA 114325 C2 5 10 15 20 25 30 35 40 1H), 3,74-3,65 (m, 1H), 3,52 (широкий d, J=12,5 Гц, 1H), 3,11-2,99 (m, 1H), 2,15 (широкий d, J=12,4 Гц, 1H), 1,89-1,71 (m, 4H), 1,52-1,45 (m, 1H), 1,40 (d, J=6,8 Гц, 3H), 1,33 (d, J=6,8 Гц, 3H). + Приклад 9b: другий ізомер (Ізомер 2). Масс-спектр (m/z): 411 (M+H) . Фахівцеві в цій галузі буде зрозуміло, що за добре відомих у цій галузі умов хлористоводневі солі Прикладів 1-9 можна легко перетворити на відповідні вільні основи. In vitro зв'язування з людським ЕР1, ЕР2, EP3 і EP4 Мембрани hEP1 і hEP4 одержують з рекомбінантних клітин НЕК293, які стабільно експресують людські рецептори EP1 (№ депонування GenBank AY275470) або EP4 (№ депонування GenBank AY429109). Мембрани hEP2 і hEP3 одержують з клітин НЕК293, короткотривало трансфекованих плазмідами рецепторів ЕР2 (№ депонування GenBank AY275471) або EP3 (ізоформа VI: № депонування Genbank AY429108). Заморожений дебріс гомогенізують в гомогенізаційному буфері із застосуванням тефлонового/скляного гомогенізатора. Мембранний білок поділяють на аліквоти, і швидко заморожують на сухому льоду перед зберіганням при температурі -80 °C. Гомогенізаційний буфер містить 10 мМ ТрисTM HCl, рН 7,4, 250 мМ сахарози, 1 мМ ЕДТА, 0,3 мМ індометацину з Complete з додаванням ЕДТА, одержаного від компанії Roche Molecular Biochemicals (№ за каталагом 1 697 498). Значення Kd для зв'язування [3Н]-PGE2 з кожним рецептором визначають при дослідженні насиченого зв'язування або гомологічної конкуренції. Сполуки випробують в 96-лунковому форматі з використанням серії трикратних розведень для одержання 10-точкової кривої. Розбавлену сполуку інкубують з 20 мкг мембрани EP1 на лунку, 10 мкг мембрани EP2 на лунку, 1 мкг мембрани EP3 на лунку або від 10 мкг на лунку до 20 мкг мембрани EP4 на лунку 3 протягом 90 хв при температурі 25 °C в присутності від 0,3 нМ до 0,5 нМ [ H]-PGE2 (компанія Perkin-Elmer, від 118 Кюрі/ммоль до 180 Кюрі/ммоль). Реакцію зв'язування здійснюють в 200 мкл MES-буферу (10 мМ MES, рН 6,0, з КОН, 10 мМ MgCl 2 і 1 мМ ЕДТА) із застосуванням 0,5 мл глибоких полістиролових 96-лункових планшетів. Неспецифічне зв'язування обчислюють порівнянням зв'язування в присутності 2 мкМ PGE2 і за його відсутності. Згадані мембрани відфільтровують (харвестер TomTek), 4 рази промивають холодним буфером (10 мМ MES, рН 6,0, з КОН, 10 мМ MgCl2), сушать в печі при температурі 60 °C, і здійснюють кількісне визначення рівня радіоактивності як число імпульсів на хвилину (СРМ) із застосуванням детектора TopCount. Відсоток специфічного зв'язування обчислюють як відсоток зв'язування за відсутності будь-якого інгібітору, з поправкою на зв'язування в присутності 2 мкМ PGE2. Дані аналізують із застосуванням 4-параметричного нелінійного логістичного рівняння (Abase Equation 205), як то: у=(A+((B-A)/(1+((С/х)^D)))), де y – відсоток специфічного інгібування, A – нижня точка кривої; B – верхня точка кривої; С – відносна IC50, тобто концентрація, що спричинює 50 % інгібування, виходячи з діапазону даних від верхньої точки до нижньої точки; D – коефіцієнт Хілла, тобто нахил кривої. Показник Ki одержують зі значень IC50 (Ki=IC50/(1+[L]/Kd), де [L] – концентрація ліганду). При випробовуванні сполук Прикладів 1-9 за цим винаходом по суті, як описано вище, вони демонструють значення Ki для hEP4 менші за приблизно 1 мкM. Таблиця 1: In vitro зв'язування сполуки Прикладу 1 з людським EP1, EP2, EP3 і EP4 Експериментальна сполука Сполука Прикладу 1 45 50 55 hEP1, Ki (нМ) hEP2, Ki (нМ) hEP3, Ki (нМ) hEP4, Ki (нМ) >17500 1550±1860 (n=3) >14000 54±27 (n=7) Зокрема, відповідно до описаного вище методу, дані, наведені в Таблиці 1, свідчать, що сполука Прикладу 1 зв'язується з hEP4 при низьких наномольних концентраціях. Дані, наведені в Таблиці 1, також свідчать, що сполука Прикладу 1 зв'язується з hEP4 краще, ніж з hEP1, hEP2 і hEP3, що вказує на селективність відносно рецептора hEP4. In vitro функціональна антагоністична активність відносно людського ЕР4 Аналізи проводять на рекомбінантних клітинах НЕК293, які стабільно експресують людський рецептор EP4. Клітинні лінії підтримують культивуванням в середовищі DMEM з високим вмістом глюкози і гідрохлориду піридоксину (компанія Invitrogen), доповненому 10 % фетальної бичачої сироватки (FBS), 1 мМ пірувату натрію, 10 мМ HEPES, 500 мкг/мл генетицину і 2 мМ Lглутаміну. Конфлюентні культури вирощують при температурі 37 °C в атмосфері, що містить 7 5 % CO2. Клітини, суспендовані в середовищі для заморожування (FBS з 6 % ДМСО) (10 клітин на мл), збирають з використанням 2,5 % розчину трипсину-ЕДТА, і аліквоти зберігають в рідкому азоті. Безпосередньо перед проведенням аналізу клітини відтаюють в середовищі DMEM, центрифугують, і ресуспендують в цАМФ буфері. 17 UA 114325 C2 5 10 15 20 25 30 35 40 45 50 55 60 Інгібування PGE2-стимульованого продукування цАМФ антагоністами EP4 визначають за допомогою HTRF; (№ за каталогом компанії Cisbio 62AM4PEB). Аліквоту, еквівалентну 4000 клітин, інкубують з 50 мкл аналітичного буферу цАМФ в концентрації, попередньо визначеній як така, що спричинює ЕС80 (10 мг 0,188 нм розчин PGE2 від компанії Sigma, № за каталогом P5640), і антагоністами EP4, при кімнатній температурі протягом 20 хв. аналітичний буфер цАМФ містить 500 мл HBSS (збалансований сольовий розчин Хенкса), 0,1 % BSA, 20 мМ HEPES і 200 мкМ IBMX (компанія Sigma, № за каталогом I5879). Як позитивний контроль використовують CJ-042794 (4-{(1S)-1-[({5-хлор-2-[(4-фторофеніл)окси]феніл}карбоніл)аміно]етил}бензойну кислоту). Для вимірювання рівнів цАМФ кон'югат цАМФ-d2 і кон'югат анти-цАМФ-криптату в лізисному буфері інкубують з обробленими клітинами при ® кімнатній температурі протягом 1 год. Сигнал HTRF виявляють планшет-рідером EnVision (компанія Perkin-Elmer) для обчислення співвідношення флуоресценції при 665 нм та 620 нм. Необроблені дані перетворюють на кількість цАМФ (пмоль на лунку) із застосуванням стандартної кривої цАМФ, яку одержують для кожного експерименту. Дані аналізують із застосуванням 4-параметричного нелінійного логістичного рівняння (Abase Equation 205), як то: у=(A+((B-A)/(1+((С/х)^D)))), де y – відсоток специфічного інгібування, A – нижня точка кривої; B – верхня точка кривої; С – відносна IC50, тобто концентрація, що спричинює 50 % інгібування, виходячи з діапазону даних від верхньої точки до нижньої точки; D – коефіцієнт Хілла, тобто нахил кривої. Відповідно до описаного вище методу, сполуки Прикладів 1-9 за цим винаходом випробують по суті як описано вище, і вони демонструють IC50 меншу за приблизно 1 мкМ. Зокрема, відповідно до описаного вище методу в разі визначення на людському EP4 сполука Прикладу 1 має IC50 6,92,5 нМ (n=5). Це свідчить про те, що сполуки Прикладів 1-9 є потужними антагоністами людського EP4 in vitro. In vitro функціональна антагоністична активність відносно пацючого ЕР4 кДНК пацючого ЕР4 (№ депонування Genebank NM_03276) клонують у векторі pcDNA 3.1 і згодом трансфекують у клітини НЕК293 для експресії рецептора. Стабільній клон пацючого ЕР4 масштабують, після чого заморожують як банк клітин для майбутнього скринінгу сполук. Для випробування сполук-антагоністів EP4 на клітинах rEP4, заморожені клітини відтаюють, а потім ці клітини ресуспендують в аналітичному буфері цАМФ. Буфер цАМФ включає HBSS без фенолового червоного (компанія Hyclone, SH30268), доповнений 20 мМ HEPES (компанія Hyclone, SH30237), 0,1 % BSA (компанія Gibco, 15260) і 125 мкМ IBMX (компанія Sigma, I5879). Клітини висівають в 96-лункові полістиролові планшети чорного кольору з половинним ребром (компанія Costar 3694). Сполуки серійно розводять ДМСО для одержання 10-точкових кривих залежності "концентрація-ефект". Потім ці розбавлені сполуки додають до аналітичного буферу цАМФ, що містить PGE2 (компанія Cayman, 14010, в заздалегідь визначеній концентрації для одержання ЕС80), в співвідношенні ДМСО/буфер 1/100. Клітини обробляють згаданими сполуками в присутності PGE2 (концентрація ЕС80) протягом 30 хв при кімнатній температурі. Рівні цАМФ, продуковані клітинами, кількісно визначають із застосуванням аналітичного набору cAMP HTRF assay kit (компанія Cisbio, 62AM4PEC). Планшети зчитують планшет-рідером EnVision із застосуванням оптимізованого протоколу HTRF (компанія PerkinElmer). Значення IC50 обчислюють підгонкою нелінійної регресійної сигмоїдної кривої залежності "доза-ефект" GraphPad Prism (v.4). Відповідно до описаного вище методу, сполуки Прикладів 1-9 за цим винаходом випробують по суті як описано вище, і вони демонструють IC50 меншу за приблизно 1 мкМ. Зокрема, відповідно до описаного вище методу в разі визначення на пацючому EP4 сполука Прикладу 1 має IC50 15 нМ. Це свідчить про те, що сполуки Прикладів 1-9 є потужними антагоністами пацючого EP4 in vitro. In vitro антагоністична активність в цільній крові людини Вважають, що інгібуюча дія PGE2 на LPS-індуковане продукування TNFα макрофагами/моноцитами опосередковується рецепторами EP4 (дивись Murase, А. et al., Life Sciences, 82:226-232 (2008)). Здатність сполуки Прикладу 1 до звертання інгібуючої дії PGE 2 на LPS-індуковане продукування TNFα в цільній крові людини є ознакою функціональної активності. Кров відбирають у здорових донорів-добровольців у вакутейнерні пробірки для гепаринізованої крові. Донори не приймали NSAID або целекоксибу протягом 48 год. або глюкокортикоїдів протягом двох тижнів до моменту відбору крові. Вміст усіх пробірок від одного донора збирають в 50 мл конічні центрифужні пробірки Falcon, і розподіляють в 96-лункових планшетах для культури тканин (компанія Falcon, 3072) з розрахунку 98 мкл на лунку. Сполуки розбавляють 100 ДМСО, і з потроєнням додають до крові з розрахунку 1 мкл на лунку для 18 UA 114325 C2 5 10 15 20 одержання 7-точкових кривих залежності "концентрація-ефект". Кров обробляють згаданими сполуками при температурі 37 °C у зволоженій атмосфері з 5 % CO2 протягом 30 хв, після чого додають 1 мкл на лунку розчину, який містить 1 мг/мл ліпополісахариду (LPS) (компанія Sigma, 0111:В4) в 0,2 мг/мл бичачому сироватковому альбуміні (BSA)/PBS з 1 мМ розчину PGE2 (компанія Cayman, 14010), і без нього, до одержання кінцевої концентрації LPS 10 мкг/мл з 10 нМ розчину PGE2 і без нього. Планшети інкубують протягом 20-24 год. при температурі 37 °C в зволожній атмосфері з 5 % СО2. Планшети центрифугують при 1800g протягом 10 хв при температурі 22 °C на центрифузі Eppendorf 5810R. З шару клітин видаляють плазму, і переносять її на поліпропіленові планшети з V-подібним дном. Кількісне визначення рівнів TNFα в 2 мкл плазми здійснюють із застосуванням комерційно доступного імуноферментного аналізу (компанія R&D Systems, DY210) з використанням планшетів Immulon 4 HBX (компанія Thermo, 3855) і 3,3',5,5'-тетраметилбіфеніл-4,4-діаміну у якості субстрату (компанія KPL, 50-76-03). Планшети зчитують при A450-A650 планшет-рідером (VersaMax, компанія Molecular Devices) з використанням програмного забезпечення SOFTmaxPRO (v.4.3.1). Значення IC 50 обчислюють підгонкою нелінійної регресійної сигмоїдної кривої залежності "доза-ефект" GraphPad Prism (v.4). Результати виражають як середнє геометричне±середнє квадратичне відхилення; де n – число незалежних визначень. Відповідно до описаного вище методу, сполуки Прикладів 1-9 за цим винаходом випробують по суті як описано вище, і вони демонструють IC50 меншу за приблизно 1 мкМ. Зокрема, відповідно до описаного вище методу сполука Прикладу 1 має IC 50 12388 нМ (n=12). Це свідчить про те, що сполуки Прикладів 1-9 є потужними антагоністами EP4 в дослідженні індукції TNFα в людській крові. ФОРМУЛА ВИНАХОДУ 25 1. Сполука формули: CO2H NH N R O 30 R O 2 3 R 4 X , де X являє собою: H3 C , R 1 або , 1 35 40 R являє собою Н, -CN або F; 2 R являє собою Н або метил; 3 R являє собою Н; та 4 R являє собою Н, метил або етил; або 3 4 R та R , сполучені разом, утворюють циклопропілове кільце; або її фармацевтично прийнятна сіль. 2 2. Сполука за п. 1, яка відрізняється тим, що R являє собою Н. 3 4 3. Сполука за п. 1 або п. 2, яка відрізняється тим, що R являє собою Н, a R являє собою метил. 4. Сполука за будь-яким з пп. 1-3, яка відрізняється тим, що X являє собою: 19 UA 114325 C2 1 R . 5. Сполука за п. 1, яка відрізняється тим, що являє собою: CO2H NH N O CH3 O , 5 або її фармацевтично прийнятна сіль. 6. Сполука за п. 5, яка відрізняється тим, що являє собою: CO2H NH N O CH3 O , 10 або її фармацевтично прийнятна сіль. 7. Хлористоводнева сіль сполуки за п. 6, яка відрізняється тим, що являє собою: CO2H HCl NH N O CH3 O 15 20 . 8. Спосіб лікування остеоартриту в пацієнта, що включає введення пацієнтові, який потребує такого лікування, ефективної кількості сполуки або її фармацевтично прийнятної солі за пп. 1-7. 9. Спосіб лікування ревматоїдного артриту в пацієнта, що включає введення пацієнтові, який потребує такого лікування, ефективної кількості сполуки або її фармацевтично прийнятної солі за пп. 1-7. 20 UA 114325 C2 5 10 15 10. Спосіб лікування болю, пов'язаного з остеоартритом або ревматоїдним артритом у пацієнта, що включає введення пацієнтові, який потребує такого лікування, ефективної кількості сполуки або її фармацевтично прийнятної солі за пп. 1-7. 11. Сполука або її фармацевтично прийнятна сіль за пп. 1-7 для застосування в терапії. 12. Сполука або її фармацевтично прийнятна сіль за пп. 1-7 для застосування в лікуванні остеоартриту. 13. Сполука або її фармацевтично прийнятна сіль за пп. 1-7 для застосування в лікуванні ревматоїдного артриту. 14. Сполука або її фармацевтично прийнятна сіль за пп. 1-7 для застосування в лікуванні болю, пов'язаного з остеоартритом або ревматоїдним артритом. 15. Фармацевтична композиція, що містить сполуку або її фармацевтично прийнятну сіль за пп. 1-7, у поєднанні з одним або більше фармацевтично прийнятними носіями, розріджувачами або наповнювачами. 16. Фармацевтична композиція за п. 15, яка відрізняється тим, що містить один або більше інших терапевтичних агентів. Комп’ютерна верстка О. Гергіль Міністерство економічного розвитку і торгівлі України, вул. М. Грушевського, 12/2, м. Київ, 01008, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 21
ДивитисяДодаткова інформація
Автори англійськоюSchiffler, Matthew Allen, York, Jeremy Schulenburg
Автори російськоюШиффлер Мэтью Аллен, Йорк Джереми Шуленбург
МПК / Мітки
МПК: A61K 31/45, C07D 211/60
Мітки: феноксіетилпіперидинові, сполуки
Код посилання
<a href="https://ua.patents.su/23-114325-fenoksietilpiperidinovi-spoluki.html" target="_blank" rel="follow" title="База патентів України">Феноксіетилпіперидинові сполуки</a>
Піримідини як інгібітори сорбітдегідрогенази, фармацевтична композиція, що їх містить, проміжні сполуки та спосіб одержання проміжної сполуки

Номер патенту: 71951
Опубліковано: 17.01.2005
Автори: Міларі Банавара Лакшман, Маррі Джеррі Ентоні, Чу-Моєр Маргарет Юхуа, Зембровскі Уільям Джеймс
МПК: C07D 417/14, C07D 513/10, C07D 401/12, C07D 491/20, C07D 491/048, A61P 3/10, C07D 491/04, C07D 491/10, C07D 413/12, C07D 471/08, C07D 487/08, C07D 401/04, C07D 521/00, C07D 498/04, A61K 31/53, C07D 417/12, C07D 451/06, C07D 405/12, C07D 471/04, A61P 9/10, C07D 239/42, A61P 43/00, A61K 31/506, C07D 487/04, C07D 403/12, C07D 409/12, C07D 403/14, C07D 401/14, C07D 409/14, C07D 403/04, C07D 405/14, C07D 451/02, A61K 31/5377, A61K 31/517
Мітки: проміжної, композиція, фармацевтична, сорбітдегідрогенази, одержання, інгібітори, проміжні, піримідини, спосіб, містить, сполуки
Формула / Реферат:
1. Похідне піримідину формули I: , Iйого пролікарська форма або фармацевтично прийнятна сіль згаданої сполуки або згаданої пролікарської форми, де: R1 є (R)-1-гідроксіетилом; R2 є воднем; R3 є; іR9 є піримідилом або триазинілом; згаданий піримідил або...
Сполуки бензимідазолу, фармацевтичні композиції, що містять ці сполуки, та спосіб лікування чутливих до позитивної модуляції гамк-рецепторних комплексів розладів цнс

Номер патенту: 54394
Опубліковано: 17.03.2003
Автори: ФУКУДА Йосімаса, Ветьєн Франк, САСАКІ Тосіро, ТЕУБЕР Лене, УСІРОДА Осаму
МПК: A61K 31/4433, A61K 31/443, A61K 31/44, C07D 413/14, C07D 409/10, A61K 31/427, A61K 31/4439, C07D 405/10, C07D 403/10, C07D 417/14, C07D 405/14, A61P 25/04, A61K 31/506, A61P 43/00, A61P 25/00, A61K 31/00, C07D 417/10, A61K 31/4427, A61K 31/4164, A61K 31/4184, C07D 401/10, A61K 31/425, A61K 31/505, C07D 521/00, A61K 31/415, C07D 235/06
Мітки: композиції, лікування, цнс, гамк-рецепторних, позитивної, комплексів, сполуки, модуляції, спосіб, фармацевтичні, розладів, бензимідазолу, чутливих, містять
Формула / Реферат:
1. Сполука формули:або її фармацевтично придатна сіль чи оксид, де R3 - ,де кожний А, В та D - СН, або один чи два з А, В та D - N, а інші - СН;R11 - феніл, бензимідазол або моноциклічний гетероарил, кожний з яких може бути заміщеним одним або більше замісниками,...
Спосіб одержання 1-(амінометил)циклогексилоцтової кислоти та проміжні сполуки

Номер патенту: 73108
Опубліковано: 15.06.2005
Автори: Салаі Крістіна, Лендьєл Зольтанн, Гізур Тібор
МПК: C07C 227/00, C07C 229/28, C07C 205/00, C07B 61/00
Мітки: 1-(амінометил)циклогексилоцтової, сполуки, спосіб, проміжні, кислоти, одержання
Формула / Реферат:
1. Спосіб одержання 1-(амінометил)циклогексилоцтової кислоти або її фармацевтично прийнятної солі, який відрізняється тим, що здійснюютьа) перетворення алкілового ефіру циклогексилінденоцтової кислоти формули (VI) - де R2 являє собою С1-С4алкільну групу - в алкіловий ефір 1-(нітрометил)циклогексилоцтової кислоти формули (V) - де значення R2 є таким, як визначено вище - за допомогою нітрометану у присутності основи, гідролізом...
Сполуки 2,3-діарилпіразоло[1,5-в]піридазинів, спосіб їх отримання (варіанти) та фармацевтична композиція на їх основі

Номер патенту: 68358
Опубліковано: 16.08.2004
Автори: Мет'юз Нейл, Бесвік Пол, Кампбелл Ян, Нейлор Елан
МПК: C07D 231/00, A61K 31/50, C07D 237/00, C07D 487/04
Мітки: варіанти, 2,3-діарилпіразоло[1,5-в]піридазинів, композиція, фармацевтична, спосіб, сполуки, отримання, основі
Формула / Реферат:
1. Сполуки 2, 3-діарилпіразоло[1,5-В]піридазинів формули (I)(І)та їх фармацевтично прийнятні похідні, в яких:R0 – галоген, С1–6алкіл, С1–6алкоксил, С1–6алкоксил, заміщений одним чи більше атомами флуору, або О(СН2)nNR4R5, R1 та R2 незалежно один від одного вибрані з групи, яка складається з гідрогену, С1–6алкілу, С1–6алкілу, заміщеного одним чи...
Дициклічні гетероароматичні сполуки як інгібітори білкової тирозинкінази

Номер патенту: 66827
Опубліковано: 15.06.2004
Автори: Лекі Керін Елізабет, антріп Стівен Беррі, Сміт Катрін Джейн, Картер Малколм Клайв, Кокерілл Джордж Стюарт
МПК: C07D 471/04, A61P 29/00, C07D 239/94, A61K 31/519, C07D 405/14, C07D 417/14, A61K 31/517, A61P 43/00, A61P 17/06, A61P 35/00, C07D 417/04, C07D 405/04, A61K 31/4709
Мітки: сполуки, інгібітори, гетероароматичні, тирозинкінази, білкової, дициклічні
Формула / Реферат:
1. Дициклічні гетероароматичні сполуки формули (І): (І)або їх солі чи сольвати як інгібітори білкової тирозинкінази,деY - CR1, a V - N,або Y - CR1, a V - CR2,R1 - група CH3SO2CH2CH2NHCH2-Ar, причому Аr вибрано з групи, яка складається з фурану та тіазолу, кожний з яких заміщено, як варіант, одним чи двома галогенами,...
Попередній патент: Засіб зберігання із протектантами рослин і його застосування
Наступний патент: Похідні діазепінону, застосовні для лікування синдрому ламкої х-хромосоми, хвороби паркінсона або рефлюксного захворювання
Випадковий патент: Спосіб визначення розмірів коронок верхніх різців