Антагоністи гормону вивільнення лутеїнізуючого гормону з поліпшеною ефективністю та спосіб їх отримання
Номер патенту: 65531
Опубліковано: 15.04.2004
Автори: Еміг Петер-Пауль, Беккерс Томас, Кучер Бернхард, Шарпентьє Патрісіа-Марі, Кленнер Томас, Бернд Міхель







Формула / Реферат
1. Сполука загальної формули V
Ас-D-Nal(2)1-D-(pCL)Phe2-D-Pal(3)3-Ser4-Tyr5-D-Xxx6-Leu7-Arg8-Pro9-D-Ala10-NH2, (V)
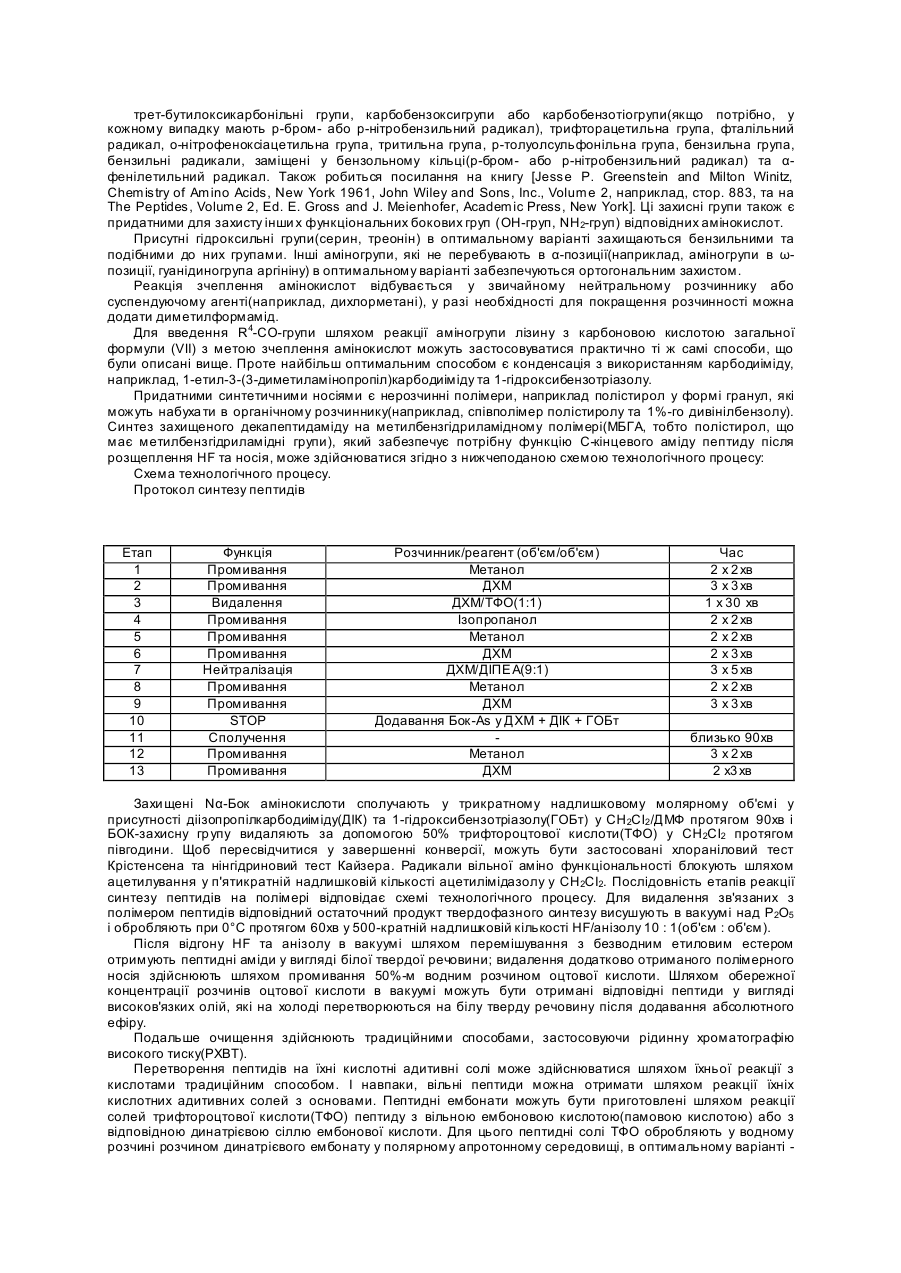
причому D-Ххх являє собою амінокислотну групу загальної формули (VI)
 ,
,
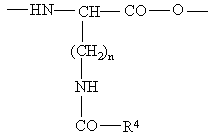
де n = 4, R4 являє собою групу формули (II)
 , (II)
, (II)
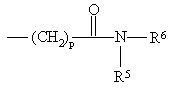
де р ціле число від 1 до 4, R5 являє собою атом водню або алкільну групу, а R6 являє собою незаміщену або заміщену арильну чи гетероарильну групу або R4 являє собою кільцеву структуру загальної формули (III)
 , (III)
, (III)
де q = 1 або 2, R7 і R8 являють собою атом водню, а Х являє собою атом сірки; та її солі з фармацевтично прийнятними кислотами.
2. Сполука за п. 1, яка відрізняється тим, що Ххх являє собою [E-N-4(4-амідинофеніл)-аміно-1,4-діоксобутил]-лізилгрупу.
3. Сполука за п. 1, яка відрізняється тим, що Ххх являє собою [E-N-(імідазолідин-2-он-4-іл)-форміл]-лізилгрупу.
4. Сполука за пп. 1-3, яка відрізняється тим, що являє собою солі ембонової кислоти.
5. Сполука загальної формули I,
 , (I)
, (I)
де n = 4, R1 - арил-, алкілокси, аралкіл-, гетероаралкіл-, аралкілокси- або гетероаралкілоксигрупа, незаміщена або заміщена, причому R2 і R3 незалежно один від одного можуть бути представлені атомом водню, алкільною групою, аралкільною групою або гетероаралкільною групою, незаміщеними або заміщеними, причому замісник, у свою чергу, може бути представлений арильною або гетероарильною групою, або –NR2R3 є амінокислотною групою, а R4 являє собою групу формули (II)
–(CH2)p–CO–NR5R6 , (II)
де р = 1-4, R5 являє собою атом водню або алкільну групу, а R6 - незаміщену або заміщену арильну або гетероарильну групу, причому замісники можуть бути представлені арильною або гетероарильною групою, або R4 являє собою кільцеву структуру загальної формули (III)
, (III)
де q= 1, 2, R7 і R8 являють собою атом водню, а X являє собою атом сірки, причому ароматичні залишки можуть бути повністю гідровані, і можуть бути представлені хіральними вуглецевими атомами з конфігурацією L; а також їхні солі з фармацевтично прийнятними кислотами.
6. Сполука за п. 5, яка відрізняється тим, що являє собою α-N-[бензилокcикарбоніл]-E-N-[5-[(4-амідинофеніл)аміно]-5-окcопентаноїл]-L-лізинамідтрифторацетат.
7. Сполука за п. 5, яка відрізняється тим, що являє собою α-N-[бензилоксикарбоніл]-E-N-[5-[(4-амідинофеніл)аміно]-4-оксобутаноїл]-L-лізинамідтрифторацетат.
8. Сполука за п. 5, яка відрізняється тим, що являє собою α-N-[бензилоксикарбоніл]-E-N-[4-[(4-амідинофеніл)аміно]-4-оксобутаноїл]-L-лізин-N-(3-піридилметил)амід-трифторацетат.
9. Сполука за пп. 5-8, яка відрізняється тим, що являє собою солі ембонової кислоти.
10. Сполука за пп. 1-9, яка відрізняється тим, що вона призначена для одержання лікарських препаратів для лікування гормонзалежних пухлин, особливо карциноми простати або раку молочної залози, а також незлоякісних пухлин, лікування яких вимагає супресії ЛГ-РФ.
11. Фармацевтична композиція, яка має високу спорідненість до рецептора рилізинг-фактора лютеїнізуючого гормону, яка відрізняється тим, що як активну речовину містить сполуки за пп. 1-4.
12. Спосіб одержання сполуки за п. 1 формули V, який відрізняється тим, що включає такі стадії:
(а) зв'язування D-аланіну, що містить блоковану амінокислоту, з придатною підкладкою для твердофазного синтезу,
(б) видалення захисної групи з аміногрупи аланіну,
(в) забезпечення реакції конденсації зв'язаного з підкладкою аланіну з проліном, який містить захисну групу на атомі азоту,
(г) видалення захисної групи з атома азоту проліну,
(д) повторення стадій (в) і (г) з амінокислотами 1-8 згідно з формулою (V), у порядку від восьмої до першої,
(е) видалення сполуки, отриманої на стадії (д), з підкладки,
(є) забезпечення реакції конденсації з карбоновою кислотою загальної формули (VII)
R4-СООН, (VII)
де R4 має значення відповідно до п. 1,
(ж) за необхідності забезпечення реакції конденсації з фармацевтично прийнятною кислотою, переважно ембоновою кислотою, з утворенням солі.
13. Спосіб одержання сполуки за п. 1 формули V, який відрізняється тим, що включає такі стадії:
(а) зв'язування D-аланіну, що містить блоковану аміногрупу, з придатною підкладкою для твердофазного синтезу,
(б) видалення захисної групи з аміногрупи аланіну,
(в) забезпечення реакції конденсації зв'язаного з підкладкою аланіну з проліном, який містить захисну групу на атомі азоту,
(г) видалення захисної групи з атома азоту проліну,
(д) повторення стадій (в) і (г) з амінокислотами 6-8 згідно з загальною формулою (V), у порядку від восьмої до шостої,
(е) видалення захисної групи з Е-аміногрупи D-лізину або D-орнітину в положенні 6 і забезпечення конденсації з карбоновою кислотою загальної формули (VII)
R4-СООН , (VII)
де R4 має значення відповідно до п. 1,
(є) видалення захисної групи з L-аміногрупи D-лізину і D-орнітину,
(ж) повторення стадій (в) і (г) з амінокислотами 1-5 згідно з загальною формулою (IV), у порядку від п'ятої до першої,
(з) видалення сполуки, отриманої на стадії (ж), з підкладки й очищення, особливо методом ЖХВД.
(і) за необхідності забезпечення реакції конденсації з фармацевтично прийнятною кислотою, переважно ембоновою кислотою, з утворенням солі.
14. Спосіб за пп. 12, 13, який відрізняється тим, що як карбонову кислоту загальної формули (VII) використовують N-(4-амідинофеніл)-аміно-4-оксоолійну кислоту.
15. Спосіб за пп. 12, 13, який відрізняється тим, що як карбонову кислоту загальної формули (VII) використовують імідазолідин-2-он-4-карбонову кислоту.
16. Спосіб за пп. 12-15, який відрізняється тим, що як фармацевтично прийнятну кислоту використовують ембонову кислоту.
17. Спосіб приготування лікарських препаратів, що містять сполуки за пп. 1-9, який відрізняється тим, що вказані сполуки змішують з допоміжними речовинами.
Текст
Даний винахід стосується нових антагоністів гормону вивільнення лютеїнізуючого гормону(ЛГ), зокрема пептидоміметиків та пептидів, модифікованих у боковому ланцюгу, їхніх солей фармацевтично прийнятних кислот та способів приготування антагоністів гормону вивільнення ЛГ та їхніх солей. Пептиди згідно з даним винаходом є аналогами гормону вивільнення лютеїнізуючого гормону, який має таку структур у: р-Глу-Пс-Трп-Сер-Тир-Глі-Лей-Арг-Про-Глі-МН 2, [гормон вивільнення ЛГ, гонадорелін]. Понад 20 років дослідники вишукували антагоністи вибірково активного гормону вивільнення ЛГ декапептиду [М. Karten and J. E Ri vier, Endocrine Reviews 1, 44 - 66 (1986)]. Великий інтерес до цих антагоністів пояснюється їхнім широким застосуванням у галузях ендокринології, гінекології, для контрацепції та лікування раку. Багато сполук були приготовлені як потенційні антагоністи гормону вивільнення ЛГ. Найбільш цікавими сполуками, виявленими на нинішній час, є сполуки, структура яких є модифікацією структури гормону вивільнення ЛГ. Першу серію потенційних антагоністів було отримано шляхом введення ароматичних естерів амінокислот у позиції 1, 2, 3 та 6 або 2, 3 та 6. Традиційно сполуки записують так: спочатку позначають амінокислоти, які вводяться у пептидний ланцюг гормону вивільнення ЛГ на тому місці, де спочатку були амінокислоти, і позиції, у яких відбувається заміщення, позначають цифрами верхнього індексу. Крім того, позначка “ГВЛГ” (гормон вивільнення лютеїнізуючого гормону), яку ставлять наприкінці, означає, що вони є аналогами гормону вивільнення ЛГ, у якому відбувається заміщення. Серед антагоністів відомими є такі: [Ac-D-Фен(4-CI)1,2 , D-Трп 3,6 ] ГВЛГ [D. H. Coy et al., In: Gross, E. and Meienhofer, J. (Eds) Peptides; Proceedings of the 6th American Peptide Symposium, pp. 775 - 779, Pierce Chem. Co., Rockville III. (1979]: [АсПро1, D-Фен (4-CI)2, D-Нал (2)3,6] ГВЛГ [US-Patent №4,419,347] та [АС-Про 1, D-Фен (4-CI)2, D-Трп 3,6 ГВЛГ [J. L Pineda, et al., J. Clin. Endocrinol. Metab. 56, 420, 1983]. Для підвищення розчинності антагоністів у воді згодом на 6 позицію вводяться основні амінокислоти, наприклад D-Apr. Наприклад, [Ас-О-Фен(4-СІ)1,2 , D-Трп 3, D-Арг6, D-Ala10] ГВЛГ [ORG-30276] [D. Н. Coy, et. al.. Endocrinology 100, 1445, 1982]; та [Ac-D-Hал(2)1, D-Фен(4-F)2, D-Трп 3, D-Apr6] ГВЛГ [ORF 18260] [J. E. Rivier et al., in: Vickery B. H. Nestor, Jr. J. J., Hafe z, E. S. E (Eds). LHRH and its Analogs, pp. 11- 22 MTP Press, Lancaster, UK 1984]. Такі аналоги мали не лише очікувану покращену водорозчинність, але також виявляли підвищену активність як антагоністи. Однак ці надзвичайно активні гідрофільні аналоги з D-Apr6 та іншими основними боковими ланцюгами у 6 позиції викликали тимчасові набряки на мордах та кінцівках щурів, коли їх вводили підшкірно у дозах 1,25 або 1,5мг/кг [F. Schmidt, et al., Contraception 29., 283, 1984: J. E Morgan, et al., Int. Archs. Allergy Appl. Immun. 80, 70 (1986)]. Решту активних антагоністів ГВЛГ описано у заявках WO 92/19651, WO 94/19370, WO 92/17025, WO 94/14841, WO 94/13313, та патентах US-A 5,300,492, US-A 5,140,009 та ЕР 0 413 209 AI. Випадки набряків у щурів після введення деяких з цих антагоністів дає привід для сумнівів щодо безпечності у разі застосування на людях, а отже, впровадження цих медикаментів для клінічного застосування відкладається. Таким чином, існує велика потреба в антагоністичних пептидах, що не мають побічних ефектів. Згідно з даним винаходом вищезгадана мета досягається через застосування сполук загальної формули (І) в якій n є числом 3 або 4, R1 являє собою алкільну груп у, алкоксигрупу, арильну груп у, гетероарильну груп у, аралкільну груп у, гетероалкільну груп у, аралкілокси групу або гетероаралкілокси групу, у кожному з випадків заміщену або незаміщену, R2 та R3 незалежно один від одного являють собою атом водню, алкільну груп у, аралкільну групу або гетероалкільну групу, у кожному з випадків незаміщену або заміщену, або -NR2R3 є амінокислотною групою, a R4 є гр упою, що має формулу (II) в якій р є цілим числом від 1 до 4, R5 є воднем або алкільною групою, a R 6 є незаміщеною або заміщеною арильною групою або гетероарильною групою, причому заміщення, у свою чергу, може відбуватися або на арильну, або гетероарильну груп у, або R4 являє собою кільце загальної формули (III) в якій q є числом 1 або 2, R7 є атомом водню або алкільною групою, R6 є атомом водню або алкільною групою, а X є киснем або атомом сірки, причому ароматичні або гетероароматичні радикали можуть бути повністю або частково гідрогенізовані, і хіральні атоми вуглецю можуть мати R- або S-конфігурацію, та їхні солі фармацевтично прийнятних кислот. Оптимальними комбінаціями радикалів від R1 до R4 є: а) R1 є бензилокси, R2 є воднем і R3 є воднем, б) R1 є бензилокси, R2 є воднем і R4 є 4-амідинофенілом, і в) R2 є воднем, R3 є воднем і R4 є 4-амідинофенілом. Оптимальними алкільними групами є метильна, етильна, n-пропільна, і-пропільна, n-бутильна, ібутильна, t-бутильна, 2-етилгексильна, додецильна та гексадецильна групи. Оптимальними арильними групами є фенільна, нафтильна, фенантренільна та флуоренільна групи. Оптимальними гетероарильними групами є піридильна, піримідильна, імідазолільна, імідазопіридильна, індолільна, індазолільна, триазолільна, тетразолільна, бензімідазолільна, хінолільна, 2,5-дихлорпірид-3-ильна та фурильна групи. Оптимальними гідрогенізованими гетероарильними групами є піперидино, піперазинільна, морфоліно та піролідинільна групи. Аралкільними групами та гетероаралкільними групами є групи, які зв'язані з відповідними зв'язуючими центрами через алкіленову гр уп у, в оптимальному варіанті - метиленову, етиленову, n-пропіленову або nбутиленову гр упи. Оптимальними замісниками є атоми галогенів, таких як фтор, хлор, бром, йод, а також метил, етил, іпропіл, трет-бутил, ціано, нітро, карбонова кислота, карбоксамід, метиловий естер карбонової кислоти, етиловий естер карбонової кислоти, етиловий естер кротонової кислоти, трифторметильна, бензоїльна, метокси, бензилокси, піридилокси, аміно, диметиламіно, ізопропіламіно, амідино та хінолілметокси групи. Крім того, згідно з винаходом, вищезгаданої мети дозволяють досягти сполуки загальної формули (V) Ac-D-Нал (2)1-D (рСІ) Фен 2-D-Пал (3)3-Сер4-Тир5-D-Ххх6-Лей7-Арг 6-Про9-D-Ала10-NН2 (V), де D-Xxx є амінокислотною групою загальної формули (VI) а n, р, q, R4, R5, R6 , R7, R8 та X є такими, як визначено вище, а також їхні солі фармацевтично прийнятних кислот. Сполуки згідно з даним винаходом мають високу антагоністичну активність і не справляють небажаних побічних ефектів, зокрема не викликають набряків. Якщо вони не є присутніми як солі слабо розчинних у воді фармацевтично прийнятних кислот, то вони додатково мають покращену водорозчинність. Крім того, сполуки мають велику спорідненість з рецептором гормону вивільнення ЛГ людини, тобто мають високу активність як інгібітори вивільнення гонадотропінів з гіпофізної залози ссавців, включаючи людину, виявляють довготривалу супресію тестостерону у щурів, а також викликають мінімальне вивільнення гістаміну in vitro. Оптимальними сполуками загальної формули (І) є: α-N-Z-[ε-N'-4-(4-амідинофеніл)аміно-1,4діоксобутил]лізинамід та α-N-Z-[ε-N'(імідазолідин-2-он-4-іл)форміл]лізинамід. Оптимальними пептидами згідно з формулою (V) є ті, у яких Ххх є [ε-N'-4-(4-амідинофеніл)аміно-1,4-діоксобутил]лізильною групою або [ε-N'-(імідазолідин-2-он-4-іл)форміл]лізильною групою. Солі фармацевтично прийнятних кислот в оптимальному варіанті є слаборозчинними у воді. Зокрема, оптимальними солями є солі 4,4'-метилен-бі(3гідрокси-2-нафтойної кислоти), відомої також під назвами ембонової або памової кислоти. Номенклатура, використовувана для визначення пептидів, відповідає номенклатурі, прийнятій Комісією з біохімічної номенклатури ІЮПАК-МБС [European J. Biochem. 1984, 138, 9 – 37], в якій аміногрупи на Nкінці традиційно зображуються зліва, а карбоксильна група на С-кінці зображується справа. Такі антагоністи ГВЛГ, як пептиди та пептидоміметики, згідно з даним винаходом включають природні та синтетичні амінокислоти, перші з яких включають Ала, Вал, Лей, Іле, Сер, Тре, Ліз, Apг, Асп, Асн, Глу, Глн, Цис, Мет, Фен, Тир, Про, Трп та Гіс. В основі скорочень для окремих амінокислотних радикалів лежать традиційні назви амінокислот: Ала - аланін, Apг - аргінін, Глі - гліцин, Лей - лейцин, Ліз - лізин. Пал - (3) 3-(3піридил)аланін, Нал - (2) 3-(2-нафтил)аланін, Фен - фенілаланін, (рСІ)Фен - 4-хлорфенілаланін, Про пролін, Сер - серин, Тре - треонін, Трп - триптофан і Тир - тирозин. Усі описані нами амінокислоти походять з L-серії, якщо інші випадки не зазначені окремо. Наприклад, D-Нал(2) є скороченням для 3-(2-нафтил)-Dаланіну, а Сер є скороченням для L-серину. Інші скорочення: Бок - трет-Бутилоксикарбоніл Боф - Бензотріазол-1-окситри-диметиламіно)фосфоній гексафторфосфат ДЦК – Дициклогексилкарбодиімід ДХМ Дихлорметан ДдZ - Диметоксифенілдиметилметиленоксикарбоніл (диметоксидиметил-Z) ДІК - Діізопропілкарбодиімід ДІПЕА - N,N-діізопропілетиламін ДМФ - Диметилформамід Фмок - Флуоренілметилоксикарбоніл HF - Рідка безводна фтористоводнева кислота ГОБт - 1-Гідроксибензотріазол РХВТ - Рідинна хроматографія високого тиску ТФО - Трифтороцтова кислота Z - Бензилоксикарбоніл Згідно з даним винаходом сполуки загальної формули (І) готують таким чином: спочатку забезпечують дві з трьох функціональних груп(α-аміно, ε-аміно та група α-карбонової кислоти) захисними групами, а потім відповідним способом здійснюють реакцію з вільною третьою функціональною групою. Якщо потрібно, то з метою досягнення кращого результату можна на першому етапі ввести проміжні захисні функціональної групи. Спеціалістам відомі придатні захисні групи та способи їх приєднання. Приклади захисних гр уп описано у роботі ["Principles of Peptide Sinthesis", Springer Verlag 1984), у книзі "Solid Phase Peptide Sinthesis" J. M. Stewart and J. D. Young, Pierce Chem. Company, Rockford, III, 1984, та у роботі Г. Barany and R. В. Merrifield "The Peptides", Ch. I, pp. 1-285, 1979, Academic Press Inc]. Синтез сполук згідно з формулою (IV) може здійснюватися або класичним способом конденсації фрагментів, або шляхом твердофазного синтезу Меррифілда з послідовною побудовою однієї фази на іншій з використанням D-лізину, вже ацильованого у боковому ланцюгу карбоновою кислотою загальної формули (VII), та реакції декапептидного елементу з відповідною карбоновою кислотою через амідний зв'язок у боковому ланцюгу D-лізину. Отже, згідно з даним винаходом існують на вибір три доступні способи приготування сполуки загальної формули (V). Перший спосіб включає етапи: (а) забезпечення групи α-аміно та карбонової кислоти D-лізину або D-орнітину відповідними захисними групами; (б) реакції D-лізину або D-орнітину з захисними групами з карбоновою кислотою загальної формули (VII) R4-COOH (VII) в якій R4 є таким, як зазначено вище; (в) видалення захисної групи з групи α-карбонової кислоти сполуки, отриманої на етапі (б) з метою введення у поз. 6 на етапі (ж), (г) сполучення D-аланіну, забезпеченого на аміногрупі захисною групою, з твердим носієм у формі смоли(синтез Меррифілда) ; (д) видалення захисної групи з аміногрупи аланіну; (e) реакції аланіну, зв'язаного з твердим носієм, з проліном, забезпеченим захисною групою на атомі азоту; (є) видалення захисної групи з атому азоту проліну; (ж) повторення етапів e) та є) з амінокислотами від 1 до 8 згідно з загальною формулою (V) у послідовності від 8 до 1 з використанням модифікованого D-лізину або D-орнітину, описаних у п. (в) в поз. 6; (з) видалення сполуки, отриманої на етапі (ж) з носія та, у разі потреби, очищення(напр. РХВТ) ; (и) у разі необхідності - реакції з фармацевтично прийнятною кислотою, в оптимальному варіанті - з ембоновою кислотою. Згідно з другим можливим способом процес приготування сполуки загальної формули (V) включає етапи: (а) сполучення D-аланіну, забезпеченого захисною групою на аміногрупі, з носієм, придатним для твердофазного синтезу; (б) видалення захисної групи з аміногрупи аланіну; (в) реакції аланіну, зв'язаного зі смолою, з проліном, забезпеченим захисною групою на атомі азоту; (г) видалення захисної групи з атому азоту проліну; (д) повторення етапів в) та г) з амінокислотами від 1 до 8 згідно з загальною формулою (V), у послідовності від 8 до 1; (е) видалення сполуки, отриманої на етапі (д) з носія; (є) реакції з карбоновою кислотою формули (VII) R4-COOH (VII) 4 у якій R є таким, як визначено вище; (ж) у разі необхідності - реакції з фармацевтично прийнятною кислотою, в оптимальному варіанті ембоновою кислотою. Третій варіант процесу приготування сполуки загальної формули (V) включає етапи: (а) сполучення D-аланіну, забезпеченого захисною групою на аміногрупі, з носієм, придатним для твердофазного синтезу; (б) видалення захисної групи з аміногрупи аланіну; (в) реакції аланіну, зв'язаного зі смолою, з проліном, забезпеченим захисною групою на атомі азоту; (г) видалення захисної групи з атому азоту проліну; (д) повторення етапів в) та г) з амінокислотами від 6 до 8 згідно з загальною формулою (V), у послідовності від 8 до 6; (е) видалення ε-аміно захисної групи з D-лізину або D-орнітину у поз. 6 та реакції з карбоновою кислотою формули (VII); R4-COOH (VII) в якій R4 є таким, як визначено вище, (є) видалення захисної групи з α-аміногрупи D-лізину або D-орнітину; (ж) повторення етапів в) та г) з амінокислотами від 1 до 5 згідно з загальною формулою (IV), у послідовності від 5 до 1; (з) видалення сполуки, отриманої на етапі (ж) зі смоли та її очи щення(напр. РХВТ); (и) у разі необхідності - реакції з фармацевтично прийнятною кислотою, в оптимальному варіанті ембоновою кислотою. Оптимальними карбоновими кислотами загальної формули (VII) є імідазолідин-2-он-4-карбонова кислота та N-(4-амідинофеніл)аміно-4-оксомасляна кислота. Сполуки формули (V) синтезують згідно з традиційними способами, наприклад, суто твердофазного або частково твердофазного синтезу, або класичним способом сполучення розчинів [див. М. Bodanszky, "Principles of Peptide Sinthesis", Springer Verlag 1984]. Наприклад, способи твердофазного синтезу описано у книзі ["Solid Phase Peptide Sinthesis" J. M. Stewart and J. D. Young, Pierce Chem. Company, Rockford, III, 1984, а також Г. Barany and R. B. Merrifield "The Peptides", Ch. I, pp. 1-285, 1979, Academic Press Inc]. Класичний синтез сполук детально описано у роботі [Metoden der Organischen Chemie(Methods of Organic Chemistry) (Houben-Weyl), Synthese von Peptide (Peptide Synthesis) E. Wunsch (Editor) 1974, Georg Thieme Verlag, Stuttgart, FRG]. Поетапний синтез здійснюють, наприклад, через першого типу ковалентне зв'язування амінокислоти з карбокси-закінченням, α-аміногрупа якої є захищеною, з нерозчинним носієм, що є традиційним для подібних цілей, видалення α-аміно захисної групи цієї амінокислоти, зв'язування наступної захищеної амінокислоти з вільною аміногрупою, отриманою таким чином за допомогою її карбоксильної групи, та зчеплення таким способом інших амінокислот пептиду для поетапного синтезування у правильній послідовності, а після зчеплення усіх амінокислот - видалення завершеного пептиду з носія і, якщо потрібно, видалення інших захисних груп з побічними функціями. Поетапну конденсацію здійснюють традиційно шляхом синтезу з відповідних амінокислот, захищених відомим способом. Можна також застосовувати автоматичні синтезатори пептидів, наприклад, типу Labortec SP 650(Bachem, Швейцарія), з використанням захищених амінокислот, які є у продажу. Серед традиційних способів зчеплення окремих амінокислот одна з одною, зокрема, застосовують такі: Спосіб симетричних ангідридів у присутності дициклогексилкарбодиіміду або діізопропілкарбодиіміду(ДЦК, ДІК). Загальний карбодиімідний спосіб. Карбодиімідно-гідроксибензотріазольний спосіб [див. The Peptides. Volume 2, Ed. E. Gross and J. Meienhofer]. Для зчеплення аргініну краще застосовувати карбодиімідний спосіб. Для інших амінокислот найчастіше застосовують спосіб симетричних ангідридів або змішаний спосіб. При сполученні фрагментів в оптимальному варіанті застосовують сполучення кислот, яке відбувається без рацемізації, або за ДЦК-1-гідроксибензотріазольним, або за ДЦК-3-гідрокси-4-оксо-3,4дигідро-1,2,3-бензотріазольним способом. Застосовують також активовані естери фрагментів. Для поетапної конденсації амінокислот особливо придатними є активовані естери N-захищених амінокислот, таких, наприклад, як N-гідроксисукцинімідні естери або 2,4,5-трихлорфенільні естери. Активними каталізаторами амінолізу є N-гідрокси сполуки, які мають приблизно ту ж саму кислотність, що й оцтова кислота, наприклад 1-гідроксибензотріазол. Доступними проміжними амінозахисними групами є групи, які можуть бути видалені шляхом гідрогенізації, наприклад бензилоксикарбонільний радикал(=Z радикал), або групи, які можуть бути видалені за допомогою слабкої кислоти. Захисними групами α-аміногрупи є, наприклад: трет-бутилоксикарбонільні групи, карбобензоксигрупи або карбобензотіогрупи(якщо потрібно, у кожному випадку мають р-бром- або р-нітробензильний радикал), трифторацетильна група, фталільний радикал, о-нітрофеноксіацетильна група, тритильна група, р-толуолсульфонільна група, бензильна група, бензильні радикали, заміщені у бензольному кільці(р-бром- або р-нітробензильний радикал) та αфенілетильний радикал. Також робиться посилання на книгу [Jesse P. Greenstein and Milton Winitz, Chemistry of Amino Acids, New York 1961, John Wiley and Sons, Inc., Volume 2, наприклад, стор. 883, та на The Peptides, Volume 2, Ed. E. Gross and J. Meienhofer, Academic Press, New York]. Ці захисні групи також є придатними для захисту інши х функціональних бокових груп (ОН-груп, NН2-груп) відповідних амінокислот. Присутні гідроксильні групи(серин, треонін) в оптимальному варіанті захищаються бензильними та подібними до них групами. Інші аміногрупи, які не перебувають в α-позиції(наприклад, аміногрупи в ωпозиції, гуанідиногрупа аргініну) в оптимальному варіанті забезпечуються ортогональним захистом. Реакція зчеплення амінокислот відбувається у звичайному нейтральному розчиннику або суспендуючому агенті(наприклад, дихлорметані), у разі необхідності для покращення розчинності можна додати диметилформамід. Для введення R4-CO-грyпи шляхом реакції аміногрупи лізину з карбоновою кислотою загальної формули (VII) з метою зчеплення амінокислот можуть застосовуватися практично ті ж самі способи, що були описані вище. Проте найбільш оптимальним способом є конденсація з використанням карбодиіміду, наприклад, 1-етил-3-(3-диметиламінопропіл)карбодиіміду та 1-гідроксибензотріазолу. Придатними синтетичними носіями є нерозчинні полімери, наприклад полістирол у формі гранул, які можуть набуха ти в органічному розчиннику(наприклад, співполімер полістиролу та 1%-го дивінілбензолу). Синтез захищеного декапептидаміду на метилбензгідриламідному полімері(МБГА, тобто полістирол, що має метилбензгідриламідні групи), який забезпечує потрібну функцію С-кінцевого аміду пептиду після розщеплення HF та носія, може здійснюватися згідно з нижчеподаною схемою технологічного процесу: Схема технологічного процесу. Протокол синтезу пептидів Етап 1 2 3 4 5 6 7 8 9 10 11 12 13 Функція Промивання Промивання Видалення Промивання Промивання Промивання Нейтралізація Промивання Промивання STOP Сполучення Промивання Промивання Розчинник/реагент (об'єм/об'єм) Метанол ДХМ ДХМ/ТФО(1:1) Ізопропанол Метанол ДХМ ДХМ/ДІПЕА(9:1) Метанол ДХМ Додавання Бок-As у ДХМ + ДІК + ГОБт Метанол ДХМ Час 2 х 2 хв 3 х 3 хв 1 х 30 хв 2 х 2 хв 2 х 2 хв 2 x 3 хв 3 х 5 хв 2 х 2 хв 3 х 3 хв близько 90хв 3 х 2 хв 2 x3 хв Захи щені Nα-Бок амінокислоти сполучають у трикратному надлишковому молярному об'ємі у присутності діізопропілкарбодиіміду(ДІК) та 1-гідроксибензотріазолу(ГОБт) у СН 2СІ2 /ДМФ протягом 90хв і БОК-захисну гр упу видаляють за допомогою 50% трифтороцтової кислоти(ТФО) у СН2СІ2 протягом півгодини. Щоб пересвідчитися у завершенні конверсії, можуть бути застосовані хлораніловий тест Крістенсена та нінгідриновий тест Кайзера. Радикали вільної аміно функціональності блокують шляхом ацетилування у п'ятикратній надлишковій кількості ацетилімідазолу у СН 2СІ2. Послідовність етапів реакції синтезу пептидів на полімері відповідає схемі технологічного процесу. Для видалення зв'язаних з полімером пептидів відповідний остаточний продукт твердофазного синтезу висушують в вакуумі над Р2О5 і обробляють при 0°С протягом 60хв у 500-кратній надлишковій кількості HF/анізолу 10 : 1(об'єм : об'єм). Після відгону HF та анізолу в вакуумі шляхом перемішування з безводним етиловим естером отримують пептидні аміди у вигляді білої твердої речовини; видалення додатково отриманого полімерного носія здійснюють шляхом промивання 50%-м водним розчином оцтової кислоти. Шляхом обережної концентрації розчинів оцтової кислоти в вакуумі можуть бути отримані відповідні пептиди у вигляді високов'язких олій, які на холоді перетворюються на білу тверду речовину після додавання абсолютного ефіру. Подальше очищення здійснюють традиційними способами, застосовуючи рідинну хроматографію високого тиску(РХВТ). Перетворення пептидів на їхні кислотні адитивні солі може здійснюватися шляхом їхньої реакції з кислотами традиційним способом. І навпаки, вільні пептиди можна отримати шляхом реакції їхніх кислотних адитивних солей з основами. Пептидні ембонати можуть бути приготовлені шляхом реакції солей трифтороцтової кислоти(ТФО) пептиду з вільною ембоновою кислотою(памовою кислотою) або з відповідною динатрієвою сіллю ембонової кислоти. Для цього пептидні солі ТФО обробляють у водному розчині розчином динатрієвого ембонату у полярному апротонному середовищі, в оптимальному варіанті у диметилацетаміді, і утворений блідо-жовтий осад відокремлюють. Подані нижче приклади ілюструють винахід, проте вони не є вичерпними. Приклад 1 Ас-D-Нал(2)-D(рСІ)Фен-D-Пал(3)-Сер-Тир-D-[ε-N'-(імідазолідин-2-он-4-іл)форміл]-Ліз-Лей-Арг-Про-DАла-NН2 Синтез здійснювали згідно зі схемою технологічного процесу на 5г полімеру mВНА(трет-бутил-4метоксфенол) (щільність наповнення 1,08ммоль/г). Лізинове сполучення утворювали у вигляді Фмок-DЛіз(Бок)-ОН й ацетилюють його імідазолідин-2-он-4-карбоновою кислотою у трикратній надмірній кількості після видалення Бок-групи у боковому ланцюгу. Після видалення захисної групи Фмок за допомогою 20% піперидину/ДМФ на N-кінці здійснювали видовження ланцюга згідно зі схемою технологічного процесу. Після видалення полімерного носія отримували 5,2г неочищеного пептиду, який очищали традиційними способами РХВТ. Після наступного висушування виморожуванням було отримано 2,1г РХВТ-гомогенного продукту емпіричної формули C74H97N18O15CI, що мав правильну FAB-MC 1514(М+Н+) (підрах. 1512,7) та відповідний спектр 1Н-ЯМР. 1 Н-ЯМР (500МГц, ДМСО-d6 , δ у млн -1): 8,56, m, 2Н, аром. Н; 8,08, m, 1Н, аром. Н; 7,81, m, 1Н, аром. Н; 7,73 m, 2Н, аром. Н; 7,66, m, 1Н, аром. Н; 7,60, s, 1Н, аром. Н; 7,44, m, 2Н, аром. Н; 7,30, d, 1H, аром. Н; 7,25 та 7,18, 2d, 2 х 2Н, аром. Н р-СІ-Фен; 6,97 та 6,60, 2d, 2 х 2Н, аром. Н Тир; 9,2-6,3, кілька сигналів, амід NH; 4,8-4,0, кілька m, Сα-Н та аліф. Н; 2,1-1,1, кілька m, залишков. аліфат. Н; 1,70, s, 3Н, ацетил; 1,22, d, 3Н, Cβ-H Ала; 0,85, dd, 6H, С δ-Н Лей Приклад 2 Ас-D-Нал(2)-D(рСІ)Фен-D-Пал(3)-Cep-Tиp-D-[ε-N'-4-(4-амідинофеніл)аміно-1,4-діоксобутил]-Ліз-ЛейАрг-Про-В-Ала-ТН 2 Здійснювали реакцію 0,7ммоль(1,03г) декапептиду Ас-D-Нал-D-(рСІ)Фен-D-Пал-Сер-Тир-D-Ліз-Лей-АргПро-D-Ала-NН2 з 1,0ммоль(0,27г) (4-амідинофеніл)аміно-4-оксомасляної кислоти у присутності 1,0ммоль(0,16г) 1-етил-3-(3-диметиламінопропіл)карбодиіміду та 1,0ммоль (0,16г) 1-гідроксибензотріазолу у свіжодистильованому ДМФ. Розчинник видаляли через 24 години в вакуумі, отриманий залишок розчиняли у воді і розчин сушили виморожуванням. Отриманий неочищений продукт реакції(1,63г) очищали за допомогою РХВТ з оберненою фазою; отримували загальну кількість(0,61г) РХВТ-гомогенного продукту емпіричної формули C81H104N19O 15CI, що мав правильну FAB-MC: 1618,7(М+Н+) (підрах. 1617,7) та відповідний спектр 1Н-ЯМР. 1 Н-ЯМР (500МГц, ДМСО-d6 , δ у млн -1): 10,4, s, 1Н та 9,15, s, 2Н та 8,8, s, 1Н, NH 4-амідиноаніліну; 8,60, m, 2H, аром. Н; 8,20, m, 1Н, аром. Н; 7,80, m, 1Н, аром. Н; 7,73, m, аром. Н; 7,61, s, 1Н, аром. Н; 7,44, m, 2Н, аром. Н; 7,30, d, ІН, аром. Н; 7,25 та 7,20, 2d, 4H, аром. Н(рСІ)Фен; 7,0 та 6,6, 2D, 4Н, аром. Н Тир; 8,3 - 7,2, кілька сигналів, амід-NH; 4,73 - 4,2, кілька мультиплетів, Сα-Н; 4,13, m, 1Н, Сα-Н; Ала; 3,78 - 2,4, кілька мультиплетів, Cβ-H та аліфат. Н; 1,72, s, 3Н, ацетил; 1,22, d, 3Н, Cβ Ала; 0,85, dd, 6Н, Сδ Лей Приклад 3 0,5г(0,3ммоль) пептидного антагоніста ГВЛГ згідно з Прикладом 1, розведеного у 50мл Н2О, перетворювали шляхом реакції з 0,130г(0,3ммоль) динатрієвого памоату у 2мл водного розчину на пептидний ембонат, який швидко відкладався у розчині у вигляді жовтого осаду. Отримували 0,281г дрібнокристалічного жовто-зеленого порошку, вміст ембонової кислоти - 33%. Приклад 4 0,3г(0,17ммоль) пептидного антагоніста ГВЛГ згідно з Прикладом 2, розчиненого у 5мл диметилацетаміду, перетворювали шляхом реакції з 0,195г(0,45ммоль) динатрієвого памоату у 2мл водного розчину на пептидний ембонат, який після додавання 50мл Н 2О отримували у вигляді жовтого осаду. Отримували 0,330г дрiбнокристалічного продукту жовтого кольору, вміст ембонової кислоти - 20%. Сполуки загальної формули І можна отримати згідно з поданими нижче Схемами 1, 3, 4 та 5, з систематичним чергуванням трьох функціональних груп R1 , R2 та R4. Схема 1 показує синтез сполуки згідно з Прикладом 1: Загальна процедура приготування сполуки загальної формули І згідно зі Схемою 1 Карбонову кислоту R4-CООH, заміщену радикалом R4, який лежить в основі загальної формули І та Схеми 1 синтезу і який у разі основного радикалу R4 також може бути присутній у вигляді солі, наприклад гідрохлориду, гідросульфату або ацетату, розчиняють або суспендують з видаленням вологи та перемішуванням у неполярному або диполярному апротонному органічному розчиннику, такому як тетрагідрофуран, діоксан, метил трет-бутиловий ефір, толуол, диметилформамід, диметилацетамід, Nметил-піролідон, диметилсульфоксид або метиленхлорид, і перемішують з основою, яка служить як кислотна пастка, наприклад з діізопропіламіном, триетиламіном, N-метилморфоліном, диметиламінопіридином або піридином. Потім додають суміш Z-(L)-лізинамідгідрохлориду у розріджувачі, причому придатним розріджувачем може бути використовуваний раніше для розведення карбонової кислоти R4-COOH, заміщеної радикалом R4. Потім рівень pH реакційної суміші регулюють з використанням однієї з основ, що застосовуються як кислотні пастки, наприклад, до pH 6,5 - 9,0, в оптимальному варіанті до 7,0 - 8,5, ще краще - до 7,0 - 7,5. Нарешті до реакційної суміші додають розчин зв'язувального реагента, напр. Бензотріазол-1-ілокси-три(диметиламіно)-фосфонійгексафторфосфату(БОФ), або бензотріазол-1ілокси-трипіролідинфосфонійгексафторфосфату(ПіБОФ), або дициклогексилкарбодиіміду(ДЦК) з подальшим перемішуванням, і через деякий час рівень pH розчину знову доводиться до вищезгаданого показника. Суспензію перемішують при температурі, наприклад, 0 - 80°С, в оптимальному варіанті - 10 50°С, ще краще - 20 - 30°С, протягом 1 - 15год., а потім відфільтровують відсмоктуванням, тверду фазу промивають і фільтрат концентрують до сухого стану в вакуумі. Залишок кристалізують шляхом розтирання з органічним розчинником, наприклад толуолом, тетрагідрофураном, ацетоном, метилетилкетоном або ізопропіловим спиртом, або очищують шляхом рекристалізації, дистиляції або колонкової або тонкошарової хроматографії на силікагелі або оксиді алюмінію. Елюентом є, наприклад, суміш метиленхлориду, метанолу та аміаку(25%) у співвідношенні 85:15:1(об'єм/об'єм) або суміш метиленхлориду, метанолу та аміаку(25%) у співвідношенні 80:25:5(об'єм/об'єм). Синтез трифторацетату: Сполуку, очищен у згідно з вищеописаною процедурою, розчиняють у протонних або апротонних розчинниках, наприклад, у спиртах, таких як метанол, ЕtOН, ізопропанол, або у циклічних ефірах, наприклад, тетрагідрофурані або діоксані, і рівень її pH доводять до 10 - 11, використовуючи 2N розчин гідроксиду натрію. Осаджену тверду фаз у відфільтровують відсмоктуванням, промивають, висушують в вакуумі й обробляють у розчині етанолу при температурі 10 - 80°С, в оптимальному варіанті – 20 - 40°С, молярним еквівалентом або 2- 4-кратною надмірною кількістю трифтороцтової кислоти. Після відстоювання розчину при 0 - 4°С протягом 24год кристалізується потрібний трифторацетат, який відфільтровують відсмоктуванням і висушують в вакуумі. Згідно з загальноюпроцедурою, яка лежить в основі Схеми 1 синтезу, сполуки синтезували способом, описаним нижче у Прикладі 5 та у Таблиці 1: Приклад 5 Трифторацетат α-N-[Бензилоксикарбоніл]-ε-N-[5-[(4-амідино-феніл)-аміно]-5-оксо-пентаноїл]-Lлізинаміду 5г(17,5ммоль) гідрохлориду 5-[[4-(аміноімінометил)феніл]-аміно]-5-оксопентанової кислоти суспендують шляхом перемішування та видалення вологи у 200мл диметилформаміду й обробляють 3,85мл(35,0ммоль) N-метилморфоліну. Додають суміш 5,53г(17,5ммоль) S-(L)-лізинамідгідрохлориду у 100мл диметилформаміду й доводять рівень pH до 7,0 - 7,5, використовуючи N-метилморфолін. Нарешті, додають розчин 9,73г(21,9ммоль) бензотріазол-1-ілокси-три(диметиламіно)фосфонійгексафторфосфату (БОФ) і через 10 - 15хв рівень pH знову доводять до 7,0 - 7,5. Суспензію жовтого кольору перемішують при постійному контролюванні рівня pH, який має бути у межах 7,0 - 7,5, протягом 3 - 4год при кімнатній температурі, безбарвний осад відфільтровують відсмоктуванням, двічі промивають диметилформамідом і фільтрат жовтого кольору випарюють до сухого стану. Маслянистий залишок випаровують за допомогою 5 х 40мл метилетилового кетону: після кожного разу 5-кратної обробки розчинником метилетилкетонову фазу виливають і видаляють. Залишковий необроблений продукт, отриманий у кристалічній формі, відфільтровують відсмоктуванням, промивають 30мл метилетилового кетону й висушують при кімнатній температурі в вакуумі. Потім тверду фазу розчиняють приблизно у 50мл етанолу й доводять pH до 10 - 11, використовуючи 2N розчин гідроксиду натрію. Осаджену основу відфільтровують відсмоктуванням, промивають водою та етанолом і висушують при 35°С в вакуумі. Вихід: 5,5г(62% від теор.) Трифторацетат: 5,5г основи обробляють при 60°С в етаноловій суспензії 5-кратною молярною кількістю трифтороцтової кислоти. Розчин залишають відстоюватися протягом доби при 4°С, отриманий трифторацетат відфільтровують відсмоктуванням і висушують при 35°С в вакуумі. Вихід: 5,9г(87,7% від теор.) Точка плавлення: 185°С Елементарний аналіз: розрах. С виявл.С 53,84 54,11 Н Н 5,65 5,74 N N 13,45 13,33 1 Н-ЯМР (500МГц ДМСО-d6, δ у млн -1): 10,47, s, 1Н, анілід, 9,14 та 8,8, 2s, NH амідин, 7,82, m, 1Н, ліз-ε-NH, 7,79 та 7,46, 2s, аромат. Н, 7,27 та 6,93, 2s, 2Н, CONH2, 7,20, d, 1Н, уретан NH, 5,0, s, 2H, бензил Н, 3,89, m, 1Н, Сα-Н, 3,0 та 2,58 та 2,40, 3m, разом 6Н, аліфат. Н, 1,60 - 1,20, 4m, разом 6Н, зал. Алі фат. Н Згідно з вищенаведеною процедурою були приготовлені інші сполуки, показані нижче в Таблиці 1, причому n всюди дорівнює 4. Точки плавлення сполук згідно з вищеподаними Прикладами вказані нижче на Таблиці 2: Таблиця 2 Точки плавлення сполук згідно з Прикладами від 5 до 34 Приклад 5 6 7 8 9 10 11 12 13 14 т. пл. °С 185 185 216 - 220 225 217 - 220 218 - 222 208 - 212 (олія) 232 - 236 194 - 198 Приклад 15 16 17 18 19 20 21 22 23 24 т. пл. °С 225 211 - 214 183 - 186 (олія) залишок у вигляді сиропу (олія) (олія) (олія) залишок у вигляді сиропу (олія) Приклад 25 26 27 28 29 30 31 32 33 34 т. пл. °С залишок у вигляді сиропу 205 - 210 172 - 177 227 - 230 225 - 229 233 - 235 215 - 218 155 (олія) (олія) Вихідні речовини сполук загальної формули І приготовляли згідно зі Схемою синтезу за Таблицею 1 Z-(L)лізинамід, який використовували як вихідну сполуку для завершального етапу синтезу за Прикладами 5 - 34, є продуктом серійного виробництва. Заміщені "арил"- або "гетероариламіно-оксоалканові кислоти", які також використовуються як вихідні матеріали за Схемою 1 синтезу, можуть бути приготовлені відомим з літератури способом за аналогією зі Схемою 2 синтезу [P. R. Bovy, Organ. Chem. 5Л 7948 (1993)]. Ароматичні або гетероароматичні аміни A-NH2, використовувані за Схемою 2 синтезу, є продуктами серійного виробництва; аміноімідазо[1,2-а]піридин, що лежить в основі сполуки за Прикладом 28, може бути синтезований за аналогією з відомими з літератури способами [R. Westwood, J. Med. Chem 3J 1098 (1988)]. "Арил"- або "гетероариламіно-оксо-алканові кислоти", вже згадані як вихідні речовини, можуть також бути приготовлені шляхом реакції амінолізу монометилалкандикарбоксилату, наприклад, монометилсуберату та монометилацелату, з ароматичним або гетероароматичним аміном у киплячому спирті, наприклад, киплячому етанолі або бутанолі, або, як варіант, в ароматичному розчиннику, наприклад, у толуолі або ксилолі, при температурі кипіння, при бажанні - в автоклаві при температурі кипіння розчинника під тиском до 50 атмосфер, концентрування реакційного розчину в вакуумі й очищення залишку шляхом кристалізації з метанолу або етанолу, або шля хом колонкової хроматографії. Як елюент може використовуватися, наприклад, суміш метиленхлориду, метанолу та аміаку(25%) у співвідношенні 85:15:1(об'єм/об'єм) або суміш метиленхлориду, метанолу та аміаку(25%) у співвідношенні 80:25:5(об'єм/об'єм). Альтернативним способом приготування сполуки загальної формули (І), де R1 є 2 3 бензилоксикарбонільною групою, a R та R є атомом водню, може бути спосіб, що складається з таких етапів: 1. Амідують груп у α-карбонової кислоти. 2. ε-аміногрупу захищають Z-групою. 3. α-аміногрупу захи щають Бок-групою, в результаті чого згодом можна вибірково видаляти амінозахисні групи. 4. Видаляють Z-гpyпy ε-аміногрупи. 5. Потрібну груп у R4-CO- вводять у ε-аміногрупу. 6. Видаляють Бок-групу на α-аміногрупі. 7. α-аміногрупу забезпечують Z-групою. Інші сполуки загальної формули І можна отримати згідно з нижчеподаною Схемою 3, де показано синтез сполуки за Прикладом 35: Загальна процедура приготування сполук загальної формули І згідно зі Схемою 3: 1-й Етап: Z-Лiз(БOK)-OH та основу, наприклад, триетиламін, діізопропіламін, N-метилморфолін, N-етилпіперидин та аліфатичний або ароматичний карбонілхлорид, наприклад, ацетилхлорид, ізобутироїлхлорид, ізовалероїлхлорид, півалоїлхлорид, бензоїлхлорид або 4-метоксибензоїлхлорид додають при температурі у межах від -30°С до 30°С, в оптимальному варіанті - від -20°С до 20°С, найкраще - від -15°С до 5°С, до диполярного апротонного або неполярного органічного розчинника, наприклад, тетрагідрофурану, ди метил сульфоксиду, диметилформаміду, ацетонітрилу, етилацетату, диметилацетаміду, Nметилпіролідону, діоксану, толуолу, ефіру, метиленхлориду або хлороформу. Через деякий час, скажімо, від 30хв до 3год, при інтенсивному помішуванні додають охолоджені до -10°С розчин або суспензію аміну у диполярному апротонному або неполярному органічному розчиннику, наприклад тетрагідрофурані, диметилсульфоксиді, диметилформаміді, ацетонітрилі, етилацетаті, диметилацетаміді, N-метилпіролідоні, діоксані, толуолі, ефірі, метиленхлориді або.хлороформі. Суспензію перемішують при температурі у межах від -30°С до 30°С, в оптимальному варіанті - від -20°С та 20°С, найкраще - від -15°С до 5°С, протягом 1 - 2 годин. По завершенні реакції основу відфільтровують відсмоктуванням під час концентрування гідрохлориду та розчинника. Маслянистий залишок обробляють апротонним або неполярним органічним розчинником, наприклад, ефіром, діізопропіловим ефіром, метил-трет-бутиловим ефіром, петролейним ефіром, толуолом, ксилолом, пентаном, гексаном. Розчин перемішують протягом деякого часу, скажімо, від 30хв до 3год, доти, доки не утвориться білий осад. Осад відфільтровують відсмоктуванням й висушують. 2-й Етап Z-Ліз(Бок)амід, отриманий згідно з вищеописаною процедурою 1-го Етапу розводять у трифтороцтовій кислоті при температурі від -20°С до 30°С, в оптимальному варіанті - від 10°С до 20°С, найкраще - від -5°С до 5°С і перемішують від 15хв до 1год. Надмірну трифтороцтову кислоту концентрують і маслянистий залишок обробляють диполярним апротонним або неполярним органічним розчинником, наприклад, диметилформамідом, метиленхлоридом, тетрагідрофураном, ацетонітрилом, N-метилпіролідоном, етилацетатом. Потім додають потрібну кислоту, основу, наприклад діізопропілетиламін, N-метилморфолін та відповідний сполучний реагент, наприклад БОФ, ПіБОФ, ДЦК у диполярному апротонному або неполярному органічному розчиннику, наприклад, диметилформаміді, метиленхлориді, тетрагідрофурані, ацетонітрилі, N-метилпіролідоні, етилацетаті. Реакція відбувається при температурі від -10°С до 100°С, в оптимальному варіанті - від 0°С до 80°С, найкраще - від 10°С до 35°С. Після реакції, що триває від 1 до 5год, та відстоювання при кімнатній температурі протягом 24год розчинник концентрують. Залишок осаджують, використовуючи органічний розчинник, наприклад воду, ізопропанол, метиленхлорид або ефір. Необроблений продукт очищають шля хом хроматографії на колонці з силікагелем. Згідно з цією загальною процедурою для Етапів 1 та 2, яка лежить в основі Схеми 3 синтезу, сполуки синтезували так, як показано нижче у Таблиці 3, причому n всюди дорівнює 4. Приклад 35 N-α-N-Z-[ε-N-4-(4-Амідинофеніл)-аміно-1,4-діоксобутил]лізин-N-(3-піридилметил))амід 1-й Етап Z-Ліз(Бок)-N-(3-піридилметил)амід N-(α-N-Z-ε-N-трет-бутилоксикарбоніл)лізин-N-(3-піридилметил)амід 4г(10ммоль) Z-Ліз(Бок)-ОН, що є продуктом серійного виробництва, 1г(10ммоль) триетиламіну та 1,26г(10ммоль) півалоїлхлориду додають при -15°С до 60мл тетрагідрофурану. Через 30хв при інтенсивному перемішуванні додавали попередньо охолоджений до -10°С розчин 1,08г(10ммоль) 3(амінометил)піридину у 20мл тетрагідрофурану. С успензію перемішують при -15°С від 1 до 2год. Триетиламінгідрохлорид відфільтровують відсмоктуванням при низькій температурі, а потім випаровують тетрагідрофуран. Маслянистий залишок обробляють 100мл діетилового ефіру. Розчин перемішують до утворення осаду у вигляді білого порошку. Осад відфільтровують відсмоктуванням й висушують. Вихід: 4г(85% від теоретичного). 2-й Етап N-(α-N-Z-[ε-N-4-(4-Амідинофеніл)-аміно-1,4-діоксобутил]лізин-N-(3-піридилметил)амід 2г(4,25ммоль) Z-Ліз(Бок)-N-(3-піридилметил)аміду розчиняють у 20мл ТФО при 0°С і розчин перемішують протягом 20хв. Надлишкову ТФО концентрують і маслянистий залишок обробляють 10мл ДМФ. Потім додають 4,6мл(42,5ммоль) N-метилморфоліну, 1,15г гідрохлориду(4,25ммоль) 4-[[4аміноімінометил) феніл]аміно]-4-оксомасляної кислоти, 2,35г(5,3ммоль) БОФ та 20мл ДМФ. С уміш перемішують при кімнатній температурі протягом 24год. ДМФ концентрують і залишок двічі варять у 40мл води, потім відфільтровують відсмоктуванням і висушують. Необроблений продукт очищають шляхом хроматографії на колонці з силікагелем, використовуючи елюент 89b(70% НССІ3, 40% МеОН, 10% CH3COONa+ в 1моль на літр NH4OH 25%). Вихід: 340мг(14% від теоретичного) Приклади від 36 до 55 отримували за аналогією з Прикладом 35. Таблиця 4 Точки плавлення(m. р.) сполук згідно з Прикладами 35 – 55 Приклад 31 32 33 34 m.р. [°С] 190-198 218-220 209 195 Приклад 38 39 40 41 m.р.[°С] 190 198 213 Приклад 45 46 47 48 m. р.[°С] 189 197 35 36 37 189-191 215-220 183 42 43 44 175 196 217 49 50 51 194 Інші сполуки загальної формули І приготовляли згідно з нижчеподаними Схемами 4 та 5. 1. Ацилування за допомогою карбонових кислот або естерів хлормурашиної кислоти згідно зі Схемами 4 та 5: При кімнатній температурі здійснюють реакцію H-Ліз(Бок)-NH2 в апротонному розчиннику(ДМФ, ДМСО) у присутності основи(ДІПЕА, Н ММ) та сполучного реагента(ДЦК, ДІК, ЕДХІ) з карбоновою кислотою для отримання аміду. Після видалення розчинника залишок обробляють водою і слаборозчинний необроблений продукт відфільтровують відсмоктуванням. Продукт може бути очищений шляхом кристалізації зі спирту(МеОН, ЕtOН < 2-РrOН) або естерів(МЕК, ЕА). Реакція Н-Ліз(Бок)-NН2 з карбонілхлоридами у водно-лужному розчині(середовище ШоттенаБауманна) дозволяє отримати потрібні похідні у вихідній кількості 90-95%. Необроблений продукт виділяють за допомогою відфільтровування відсмоктуванням й очищають шляхом рекристалізації зі спирту (МеОН/ЕtOН/ізопропанол) або етилацетату чи метилетилкетону. 2. Видалення Бок-захисної групи з використанням ТФО: Видалення Бок-захисної групи при кімнатній температурі у суміші дихлорометану та три фтороцтової кислоти (2:1) піддається вимірюванню приблизно через 60хв. Виділений, як правило, маслянистий необроблений продукт R1-Ліз-NH2 швидко вступає у реакцію без подальшого очищення. 3. Ацилування при R4 = гідрохлорид 4-((4-(аміноімінометил)-феніл)аміно)-4-оксомасляної кислоти: Реакцію з іншою карбоновою кислотою(R4) здійснюють в апротонних розчинниках(ДМФ, ДМСО) при кімнатній температурі у присутності основи(НММ, ДІПЕА) з використанням сполучних реагентів, таких як ЕДХІ, Боф або ПІБоф. Після видалення розчинників продукт осідає, якщо додати води. Очищення здійснюють за допомогою РХВТ на колонці RP18. з використанням як елюенту сумішей води, ацетонітрилу та трифтороцтової кислоти. Продукт отримують у вигляді солі ТФО. Згідно з цією загальною процедурою, на якій базуються Схеми 4 та 5 синтезу, сполуки синтезували способом, описаним нижче у Прикладі 56 та Таблиці 5: Приклад 56 32ммоль Z-лізинамідгідрохлориду та 32ммоль гідрохлориду 4-((4-(аміноімінометил)феніл)аміно)-4оксимаслямої кислоти додають при кімнатній температурі до 120мл сухого дегазованого N.Nдиметилформаміду(ДМФ). Вихідні матеріали швидко розчиняють шляхом перемішування; після додавання 104ммоль діізопропілетиламіну та 40ммоль БОФ суміш перемішують при кімнатній температурі протягом 16год. Розчинник та надлишок ДІПЕА відпарюють на роторному випарнику при температурі 50-55°С та тиску близько 10мбар. Маслянистий залишок обробляють 250мл води, гомогенізують в ультразвуковій ванні й охолоджують. Осаджений необроблений продукт відфільтровують через усотування й промивають водою на капілярному фільтрі. Після висушування в вакуумі над хлоридом кальцію отримують приблизно 16г бежевого порошку, що має чистоту близько 90%(РХВТ), у вигляді солі НСІ. Для приготування відповідного трифторацетату продукт суспендують у 100мл води й обробляють 32ммоль(2,45мл) трифтороцтової кислоти(99%). Для видалення надлишкової кислоти суміш швидко видаляють на роторному випарнику, а потім ліофілізують водну суспензію. Після рекристалізації зі спирту(ЕtOН/МеОН) отриманий таким чином продукт може бути ще раз ліофілізований для покращення його розчинності. Вихід: 5,26г Т. пл.: 210-213°С Таблиця 6 Точки плавлення сполук згідно з Прикладами 56-82 Приклад 56 57 Т. пл [°С] 210-213 220-223 Приклад 65 66 Т. пл [°С] 218-222 216-219 Приклад 74 75 Т. пл [°С] 191-193 186-188 58 59 60 61 62 63 64 213-215 223-226 up to 233 up to 237 up to 221 up to 220 230-236 61 68 69 70 71 72 73 235-238 up to 218 205-208 168-170 197-202 221-226 225-228 76 77 78 79 80 81 82 220-222 210-215 up to 223 up to 226 194-197 215-222 219-222 Примітка: "up to..." означає, що речовина, яка утворює аморфну піну, має відповідні фізичні властивості після висушування виморожуванням. Точки плавлення як такої не існує; існує скоріше повільне спікання до розрідження. Сполуки згідно з даним винаходом можуть також бути присутні у формі кислотних адитивних солей, наприклад солей мінеральних кислот, наприклад соляної кислоти, сірчаної кислоти, фосфорної кислоти, солей органічних кислот, наприклад оцтової кислоти, трифтороцтової кислоти, молочної кислоти, малонової кислоти, малеїнової кислоти, фумарової кислоти, глкжонової кислоти, глюкуронової кислоти, лимонної кислоти, ембонової кислоти, метансульфонової кислоти, гідроксиетансульфонової кислоти, піровиноградної кислоти та бур штинової кислоти. І сполуки загальної формули І, і їхні солі є біологічно активними. Сполуки загальної формули І можуть призначатися у вільній формі або у формі солей фізіологічно прийнятних кислот. Вони можуть призначатися перорально, парентерально, внутрішньовенно, трансдермально або для інгалляції. Винахід також стосується фармацевтичних препаратів, що містять принаймні одну сполуку формули І або її сіль фізіологічно прийнятної неорганічної або органічної кислоти та, у разі потреби, використовувані у фармацевтиці ексципієнти та/або розріджувачі або допоміжні речовини. Приклад 83 Зв'язуюча спорідненість Cetrorelix, Прикладу 1, Прикладу 2 та Прикладу 56 з рецептором ГВЛГ людини. (Cetrorelix: Ас-D-Нал(2)-D-р-СІ-Фен-D-Пал(3)-Сер-Тир-D-Цит-Лей-Арг-Про-D-Ала-NН2) Спосіб визначення зв'язуючої спорідненості(константа диссоціації Кд): Зв'язуючу послідовність визначали за допомогою тесту конкурентного зв'язування("displacement binding experiment"; Beckers et al. Eur. J. Biochem. 231, 535-543, 1995). Як радіомічений ліганд використовується [125 І] Cetrorelix(специфічна активність 5-10 х 105 чрх/пмоль(число розпадів на хвилину/пмоль), розчинений у 20%(об'єм: об'єм) ацетонітрилу, 0,2%(маса: об'єм) альбуміну, 0,1%(маса: об'єм) ТФО, »80%(об'єм: об'єм) водний розчин). Зв'язуюча здатність йодованого пептиду становить від 60% до 85%. Як немічені тестові сполуки використовувалися Cetrorelix, Приклад 1, Приклад 2 та Приклад 5 у розчині. Речовини використовувалися у концентраціях 0,01нМ - 1000нМ(Cetrorelix, Приклад 1, Приклад 2) або 0,01мкМ-10мкМ(Приклад 56). Клітини клону окремої клітини L3.5/78, що переекспресовує рецептор ГВЛГ людини, які використовуються для тесту зв’язування, видаляють за допомогою ПБС/ЕДТА(ПБС без Са 2+/Мg2+/1нМ ЕДТА) з чашки з клітинами культури, які розвивалися в однорідних умовах; визначають число клітин і клітини ресуспендують в інкубаційному середовищі(Модифіковане середовище Ігла, Dulbecco з 4,5г/л глюкози, 10мм Hepes, pH 7,5, 0,5%(маса/об'єм) БСА, 1г/л бацитрацину, 0,1г/л SBTI, 0,1%(маса/об'єм) NaN3) при відповідній щільності клітин. Спочатку у спеціальні посудини для реакції по 400мкл(типу РеннераБекмана) вводять 200мкл масляної суміші силікон/парафін(84/16% за об'ємом) і зверху піпеткою додають 50мкл клітинної суспензії(2,5 х 105 клітин). До клітинної суспензії на масляному шарі силікону/парафіну додають 50мкл зв'язуючого середовища, що містить [125І] Cetrorelix та сполуку, призначену для тестування. Потім суміш інкубують з обертанням протягом 60хв при 37°С у термокамері. Після цього етапу, її центрифугують при 9000об/хв(при кімнатній температурі) протягом 2хв у центрифузі Heraeus Biofuge 15 з ротором HTA 13,8. В ході цієї операції клітини гранулюються крізь масляний силіконово-парафіновий шар і таким чином відокремлюються від зв'язуючого середовища. Після центрифугування реакційну посудину заморожують у рідкому азоті і щипцями знімають верхівку з вмісту реакційної посудини(клітинний осад), а потім цю верхівку, що містить клітинний осад(зв'язаний ліганд [125І] Cetrorelix) та супернатант(незв'язаний, вільний ліганд [125І] Cetrorelix) переносять у мірні пробірки. Для визначення максимального зв'язування(Во) не треба додавати конкуренту. Для визначення неспецифічного зв'язування для конкуренції додають 1мкМ неміченого Cetrorelix. При £10% від загального зв'язування Во неспецифічне зв'язування є низьким. Кількісну оцінку здійснюють у g-лічильнику; аналіз здійснюють, використовуючи програму EBDA/ligand V3.0(McPherson, J. Pharmacol. Methods 14, 213-228, 1985). Графік залежності "доза-реакція" дозволяє здійснити оцінку ІС 50(концентрація, яка викликає 50%-е уповільнення реакції рецептора), а програма EBDA/ligand на основі цих даних розраховує константу диссоціації Кд [нМ]. Результат: крива конкуренції(див. Фіг.1) показує, що усі тесто вані сполуки конкурують з радіоміченим лігандом [125І] Cetrorelix) для зв'язування з рецептором ГВЛГ людини. На всіх графіках показано залежність зв'язування(у % від загального зв’язування Во) від концентрації конкуренту. Для сполук показаних на фіг.1, можна розрахувати наступн у зв'язуючу спорідненість як константу диссоціації Кд [нМ]: Cetrorelix(SB-75) 0,214нМ, Приклад 1 - 0,305нМ, Приклад 2 - 0,104нМ і Приклад 56 - 986нМ. Зв'язуючу спорідненість як середню величину різних вимірювань можна взяти з Таблиці 7. Антагоністична дія Прикладу 2 та Прикладу 56 у функціональному аналізі рецептора ГВЛГ людини. Спосіб визначення ІР3(D-міо-1,3,5-трифосфат): субконфлюентну культур у клону клітини(L 3,5/78), що переекспресовує рецептор ГВЛГ людини, промивають один раз ПБС, клітини видаляють за допомогою ПБС/ЕДТА і суспензію клітин осаджують. Клітини ресуспендують в інкубаційному середовищі(модифіковане середовище Ігла Dulbecco з 4,5 г/л глюкози, 10мм Hepes, pH 7,5, 0,5%(маса/об'єм) БСА, 5мм LiCI, 1г/л бацитрацину, 0,1г/л SBTI), розподіленого на частини по 1,5мл у реакційні посудини й попередньо інкубованого при 37°С протягом 30хв. На кожну мірну позначку потрібно 4 х 106 клітин у 500мкл об'єму. Після етапу попереднього інкубування до суспензії клітин додають ГВЛГ(концентрований розчин 0,5мм у 10мм трис, pH 7,5, 1мм дитіотреїтолу, 0,1%(маса: об'єм) БСА/Bachem Art # Н4005) в остаточній концентрації 10нМ. Дію антагоніста випробують шляхом додавання у відповідній концентрації(наприклад, 0,0316, 0,1, 0,316 і т. д. до 100нМ для Прикладу 2). Як негативний контроль інкубують клітини без додавання ГВЛГ. Після інкубування при 37°С протягом 15хв утворений ІР3 виділяють з клітин за допомогою екстракції трихлороцтовою кислотою(ТХО). Для цього до суспензії клітин додають 500мкл крижаного 15%-го розчину(маса: об'єм) ТХО. Утворений в результаті осад піддають центрифугуванню при 4°С у центрифузі Heraeus Biofuge 15R на швидкості 2000г протягом 15хв. Надосадову рідину у кількості 950мкл екстрагують тричі з 10 об'ємами холодного насиченого водою діетилового ефіру у 15-мілілітровій посудині, поставленій на лід. Після останнього етапу екстрагування розчин доводять до рівня pH 7,5 за допомогою 0,5М розчину NaHCO3. Визначення концентрації ІР3 в екстрактах клітин здійснюють за допомогою чутливого тесту на конкурентне зв’язування з використанням зв’язуючого протеїну ІР3, міченого [3Н]-ІР3 та неміченого ІР3. Для цього використовують комплект для аналізу фірми Amersham(TRK 1000); визначення здійснюють, як описано у протоколі аналізу. Після здійснення кількох етапів нарешті додають 2мл сцинтилятора для водних зразків(Rotiszint Ecoplus), обережно домішують ресуспендовані гранули, що містять зв'язок [3Н]-ІР3 і вимірюють у b-сцинтиляційному лічильнику. Кількість пористого ІР3 розраховують з використанням стандартної кривої і вибудовують криву залежності "доза-реакція". ІС50 можна оцінити за точкою перегину цієї кривої. Результат: на Фігурі 1 показано відповідні криві "доза-реакція" для антагоністів пептиду за Прикладом 2(Фіг.2), а також для пептидоміметика за Прикладом 56(Фіг.3). Стимулювання здійснювали за допомогою 10нМ ГВЛГ та інгібування утворення ІР3, визначеного як функція концентрації речовини. Для прикладу 2 та Прикладу 56 було неможливо визначити активність агоністів, тобто самі по собі речовини не викликають стимулювання синтезу ІР3. У контрольних дослідах, не представлених нами, було продемонстровано, що нетрансфіковані клітини не можуть бути стимульовані ГВЛГ для синтезу ІР3. Концентрації ІР3, які ще піддаються вимірюванню при найвищих концентраціях, відповідають концентраціям нестимульованих клітин. У Прикладі 2 та Прикладі 56, таким чином, маємо справу з функціональними антагоністами ГВЛГ. Речовини, однак, мають різну активність. За різних експериментальних умов ІС 50 Прикладу 2 становить приблизно 0,4нМ, проте ІС50 Прикладу 56, становить приблизно 4мкМ. Ці показники активності дуже добре корелюються зі зв'язуючою спорідненістю in vitro, яка визначається тестом на конкурентне зв'язування з використанням [125І]-Cetrorelix, Кд = 0,109нМ для Прикладу 2 і Кд = 1,08мкМ для Прикладу 56. Приклад 85 Гормонопригнічуюча дія Прикладу 1, Прикладу 2 та Прикладу 56 у здорового самця щура. Для визначення пригнічення тестостерону в крові здорових самців щурів речовину вводили підшкірно у правий бік тварин. Доза становила 1,5мг/кг для Прикладу 1 та Прикладу 2, і 10мг/кг - для Прикладу 56. Для перевірки показників тестостерону з під’язикової вени тварин брали приблизно по 300мкл крові через 0, 2, 4, 8(лише для Прикладу 56), 24, 48, 72 та 96год, а потім - кожні 3 дні до закінчення пригнічення. Пригнічення 1нг/мл тестостерону після введення Прикладу 1 тривало до 264год в однієї тварини, до 336год у дво х тварин і до 384год в однієї тварини(Фіг.4). Після введення Прикладу 2, рівень тестостерону у однієї тварини був пригнічений до 408год, а у чотирьох тварин - до 648год(Фіг.5). Приклад 56(10мг/кг с. л.) пригнічував рівень тестостерону в усі х 5 тварин вже через 2год, і його дія тривала до 8год. При наступному вимірюванні(24год) показник тестостерону знову підвищи вся(Фіг.6). З'єднуюча спорідненість з рецептором ГВЛГ людини(виражена як константа диссоціації Кд [нМ]; оцінка з використанням програми EBDA/Ligand Analysis; показано середні значення різних експериментів; номер експерименту у дужках), а також пригнічення тестостерону in vivo, вивільнення гістаміну in vitro та водорозчинність порівняно з SB-75: Таблиця 7 Біологічні дані Речовина Спорідненість з рецептором ГВЛГ людини [нмоль/л] Cetrorelix SB-75 Приклад 1 Приклад 2 Приклад 3 Приклад 4 Приклад 56 0,202(10) 0,306(2) 0,109(2) 0,170(2) 0,206(2) 1082(2) (1,5мг/кг, одинична доза) пригнічення тестостерону у щурів(год) 144 336 648 864 696 * Не визначається через погану розчинність (IС50) Вивільнення гістаміну [мкг/мл] Розчинність в Н 2О [мг/мл] 9,7 31,9 17,1 н. в.* н. в. 9 27 23 н. в. н. в.
ДивитисяДодаткова інформація
Назва патенту англійськоюLuteinising hormone releasing hormone antagonists with an improved effiiciency and a method for the preparation thereof
Автори англійськоюBeckers Thomas
Назва патенту російськоюАнтагонисты гормона высвобождения лутеинизируючего гормона с улучшенной эффективностью и способ их получения
Автори російськоюБеккерс Томас
МПК / Мітки
МПК: A61K 31/47, A61K 31/4409, C07D 233/26, C07D 249/08, C07D 233/32, A61K 31/401, C07D 209/08, C07D 207/27, A61K 31/4402, C07K 5/06, C07D 233/64, A61P 5/00, C07D 231/56, A61K 31/4439, C07C 271/22, A61K 31/5355, C07D 487/04, A61K 31/4184, A61K 31/34, A61K 31/4427, C07D 213/80, C07D 307/52, A61K 31/4406, A61K 31/535, C07D 213/84, C07C 257/00, C07D 213/75, A61K 31/4015, C07D 233/61, C07D 215/14, C07D 213/40, A61K 31/4164, C07D 295/12, C07D 257/00, A61K 31/415, C07D 235/16, C07D 213/61, A61K 31/165, A61K 31/416, C07D 213/64, A61K 31/404, A61K 31/341, C07D 239/42, A61K 31/166, A61K 31/403, C07D 235/14, A61K 31/40, C07D 521/00, C07D 211/14, A61K 31/44, A61K 31/454, A61K 31/216, A61K 31/445, A61K 31/275, C07D 471/04, A61K 31/215, A61K 31/5375, C07K 7/23, C07D 215/22, A61K 31/00
Мітки: поліпшеною, лутеїнізуючого, вивільнення, антагоністи, отримання, спосіб, гормону, ефективністю
Код посилання
<a href="https://ua.patents.su/25-65531-antagonisti-gormonu-vivilnennya-lutenizuyuchogo-gormonu-z-polipshenoyu-efektivnistyu-ta-sposib-kh-otrimannya.html" target="_blank" rel="follow" title="База патентів України">Антагоністи гормону вивільнення лутеїнізуючого гормону з поліпшеною ефективністю та спосіб їх отримання</a>
Сполуки з властивостями вивільнення гормону росту, фармацевтична композиція, спосіб стимулювання вивільнення гормону росту з гіпофізу і спосіб підвищення швидкості росту тварин

Номер патенту: 61056
Опубліковано: 17.11.2003
Автори: ТЬОГЕРСЕН Хеннінг, МАДСЕН Келль, Пешке Бернд, ЛУНДТ Бехренд Фрідріх, ХАНСЕН Біргіт Сехестед, ЙОХАНСЕН Нільс Лангеланд, Лау Йєспер, ХАНСЕН Томас Крузе
МПК: A61K 38/00, C07K 5/065, A61P 15/00, A61P 13/02, C07K 7/06, C07K 5/087, C07K 14/60, C07C 237/22, C07K 5/083, A61P 5/00, C07K 5/103, C07K 5/062
Мітки: вивільнення, підвищення, сполуки, гіпофізу, спосіб, фармацевтична, гормону, властивостями, швидкості, композиція, стимулювання, росту, тварин
Формула / Реферат:
1. Сполука згідно з загальною формулою (1) A-B-C-D(-E)p (1) де p дорівнює 0 або 1,А являє собою водень або R1-(CH2)q-(X)r-(CH2)s-CO-, деq дорівнює 0 або цілому числу, вибраному з групи: 1,2,3,4,5, r дорівнює 0 або 1, s дорівнює 0 або цілому числу, вибраному з групи: 1,2,3,4,5,R1 являє собою водень, імідазоліл, гуанідино, піперазинo, морфоліно, піперидино або N(R2)-R3, де кожний радикал R2 та R3 незалежно...
Похідні пептиду (варіанти), що здатні вивільнювати гормон росту, фармацевтична композиція (варіанти), спосіб стимулювання вивільнення гормону росту з гіпофіза, способи підвищення швидкості і темпу росту та виро
Номер патенту: 55381
Опубліковано: 15.04.2003
Автори: Лау Йеспер, ХАНСЕН Томас Крузе, Пешке Бернд, ЙОХАНСЕН Нільс Лангеланд, Анкерсен Мікаель
МПК: A61K 31/42, C07C 237/32, C07C 237/36, A61K 31/4245, C07D 277/20, C07D 257/00, A61K 38/00, A61P 43/00, C07D 277/56, C07D 413/12, A61K 31/415, C07C 233/38, C07D 417/12, A61K 31/41, C07D 211/60, C07D 401/12, C07K 14/60, C07K 5/065, A61K 31/4465, C07C 237/22, A61K 31/165, A61K 31/426, C07D 211/62, C07D 271/06, A61K 31/4427, A61K 31/27, A61K 31/445, C07D 271/10, A61K 31/425
Мітки: здатні, виро, росту, пептиду, підвищення, похідні, способи, композиція, гормон, стимулювання, фармацевтична, спосіб, темпу, швидкості, гіпофіза, варіанти, вивільнення, вивільнювати, гормону
Формула / Реферат:
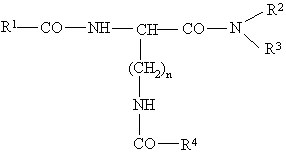
1. Похідне пептиду загальної формули I, (I)де n являє собою 0 або 1,m являє собою 1 або 2,p являє собою 0, 1 або 2,А являє собою , , , , або , де R1 - водень або С1-4-алкіл,W є =O або =S,за умови, що коли n = 1 і А - вторинний або третинний амід, або вторинний...
Спосіб одержання катіонних кристалів гормону росту, кристали гормону росту людини, фармацевтичний препарат, спосіб одержання гормону росту

Номер патенту: 26696
Опубліковано: 12.11.1999
Автори: ТЕЙСЕН Клаус Фрііс, ЮНКЕР Флемінг
МПК: C30B 7/00, C07K 1/14, C07K 14/00, A61K 38/27, C07K 14/575, C07K 14/61
Мітки: спосіб, гормону, одержання, фармацевтичний, препарат, людини, кристалів, кристали, катіонних, росту
Формула / Реферат:
1. Способ получения катионных кристаллов гормона роста или производных гормона роста, отличающийся тем, что он включает следующие стадии:а) введение в раствор гормона роста или его производного катионов неорганической или органической природы при величине pH в пределах 5,0 - 6,8;б) выращивание кристаллов при температуре примерно от 0 до 30°C;в) выделение упомянутых катионных кристаллов известными методами.2....
Аміди карбамоїлкарбоксилової кислоти, спосіб їх отримання та засіб боротьби з грибами

Номер патенту: 57000
Опубліковано: 16.06.2003
Автори: Веттеріх Франк, Лоренц Гізела, Айкен Карл, Дітріх Клаус, Вагнер Олівер, Штратманн Зігфрид, Аммерман Еберхард
МПК: A01N 47/12, A01P 3/00, C07C 271/22
Мітки: боротьби, спосіб, кислоти, засіб, карбамоїлкарбоксилової, аміди, грибами, отримання
Формула / Реферат:
1. Аміди карбамоїлкарбонової кислоти загальної формули І (І)зі ступенем чистоти ізомерів більше 90 мас.%, в яких змінні мають наступні значення:R1 означає С1-С8алкіл, С2-С8алкеніл або С2-С8алкініл, причому ці радикали можуть бути частково або повністю галогенованими і/або можуть нести від однієї до трьох груп з числа наступних: ціано, С1-С4алкокси, С1-С4галогеналкокси, С1-С4алкілтіо, С1-С4алкоксикарбоніл, С3-С7циклоалкіл,...
Антагоністи ангіотензину ii, спосіб їх отримання (варіанти), проміжна сполука (варіанти), спосіб їх отримання та фармацевтична композиція

Номер патенту: 42669
Опубліковано: 15.11.2001
Автори: Клєман Жак, Бернар Клод, Перро П'єр, Брельєр Жан-Клод, Нісато Діно
МПК: A61K 31/435, A61K 31/4184, A61K 31/415, C07D 521/00, A61P 27/02, A61P 9/04, C07D 417/12, A61P 25/00, C07D 239/70, A61K 31/445, A61K 31/454, C07D 417/10, C07D 239/52, C07D 491/107, C07D 233/70, C07D 239/60, A61P 43/00, A61K 31/496, C07D 235/02, C07D 239/36, A61K 31/505, C07D 233/84, A61K 31/425, A61P 9/12, C07D 403/10, C07D 491/10, C07D 471/10
Мітки: варіанти, ангіотензину, композиція, спосіб, сполука, проміжна, фармацевтична, отримання, антагоністи
Формула / Реферат:
1. Антагонисты ангиотензина II формулы (I), (I)где:R1 представляет собой атом водорода, карбоксигруппу, (С1-С4)алкоксикарбонильную группу, тетразолил, метилтетразолил, трифторметил-сульфониламиногруппу, трифторметилсульфониламинометил, циано, N-цианокарбамоил, N-гидроксикарбамоил, N-(4-карбокси-1,3-тиазол-2-ил)карбамоил, уреидо, 2-цианогуанидинкарбонил, имидазол-1-илкарбонил, 2-цианогуанидино-метил,...
Попередній патент: Медичний лубок і спосіб його одержання
Наступний патент: Дієтична композиція, що абсорбується, для поліпшення біологічного балансу флори кишкового тракту
Випадковий патент: Збруйне кріплення з сечозбірником (варіанти), сечозбірник та його верхня частина