Похідні 6н-дибензо[b,e]оксепіну як нестероїдні антагоністи рецепторів мінералокортикоїдів
Номер патенту: 100131
Опубліковано: 26.11.2012





Формула / Реферат
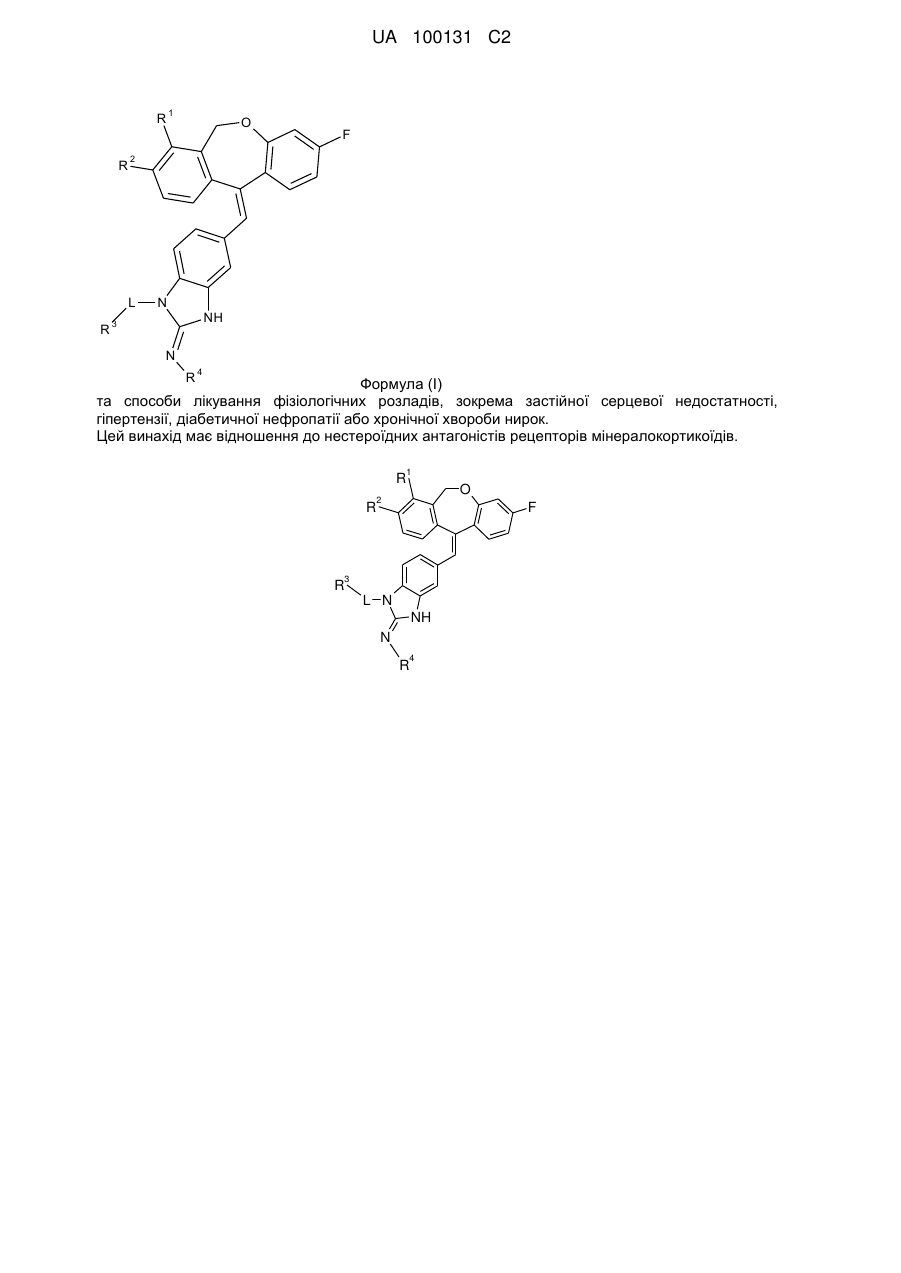
1. Сполука формули
 ,
,
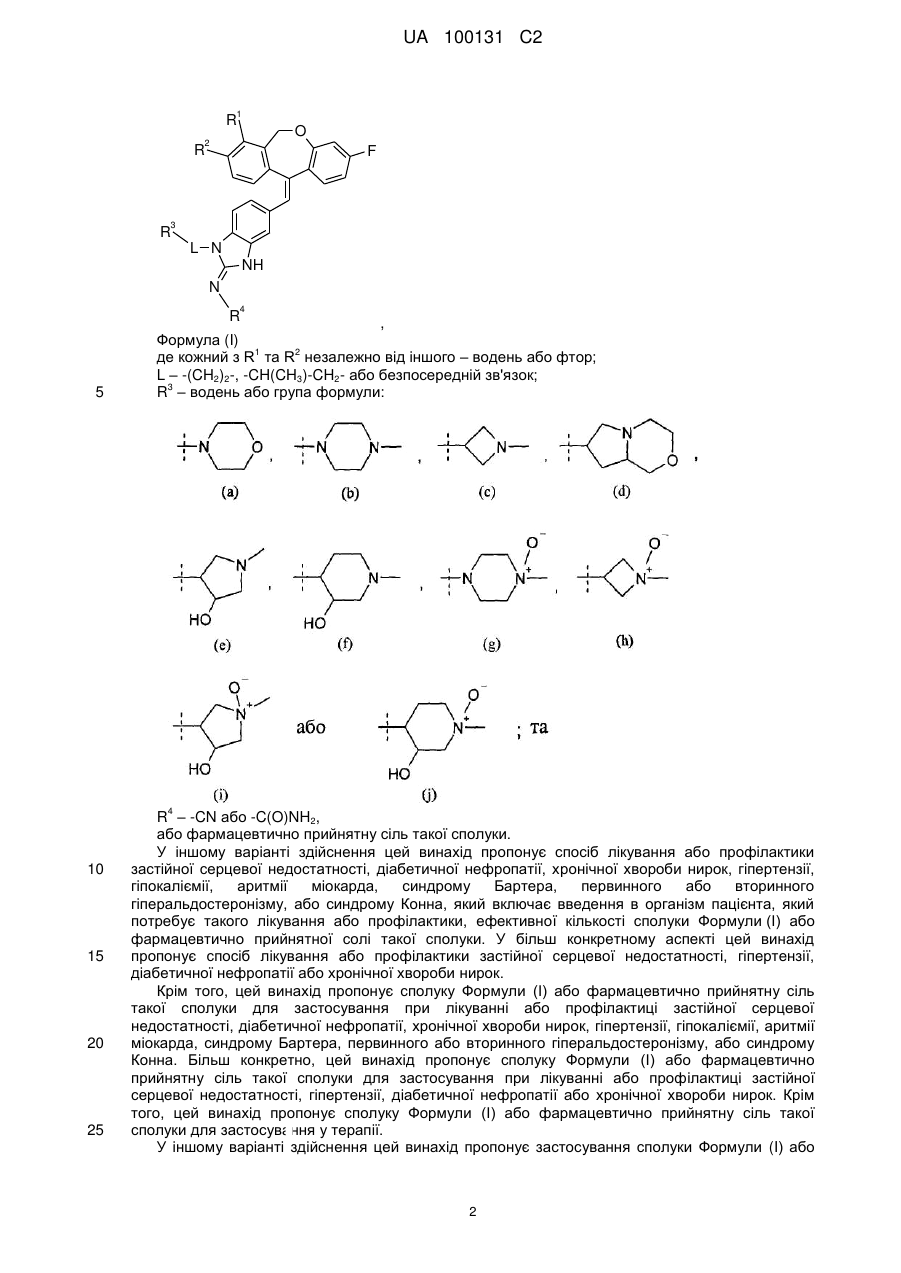
де кожний з R1 та R2 незалежно від іншого - водень або фтор;
L - група -(СН2)2-, -СН(СН3)-СН2- або безпосередній зв'язок;
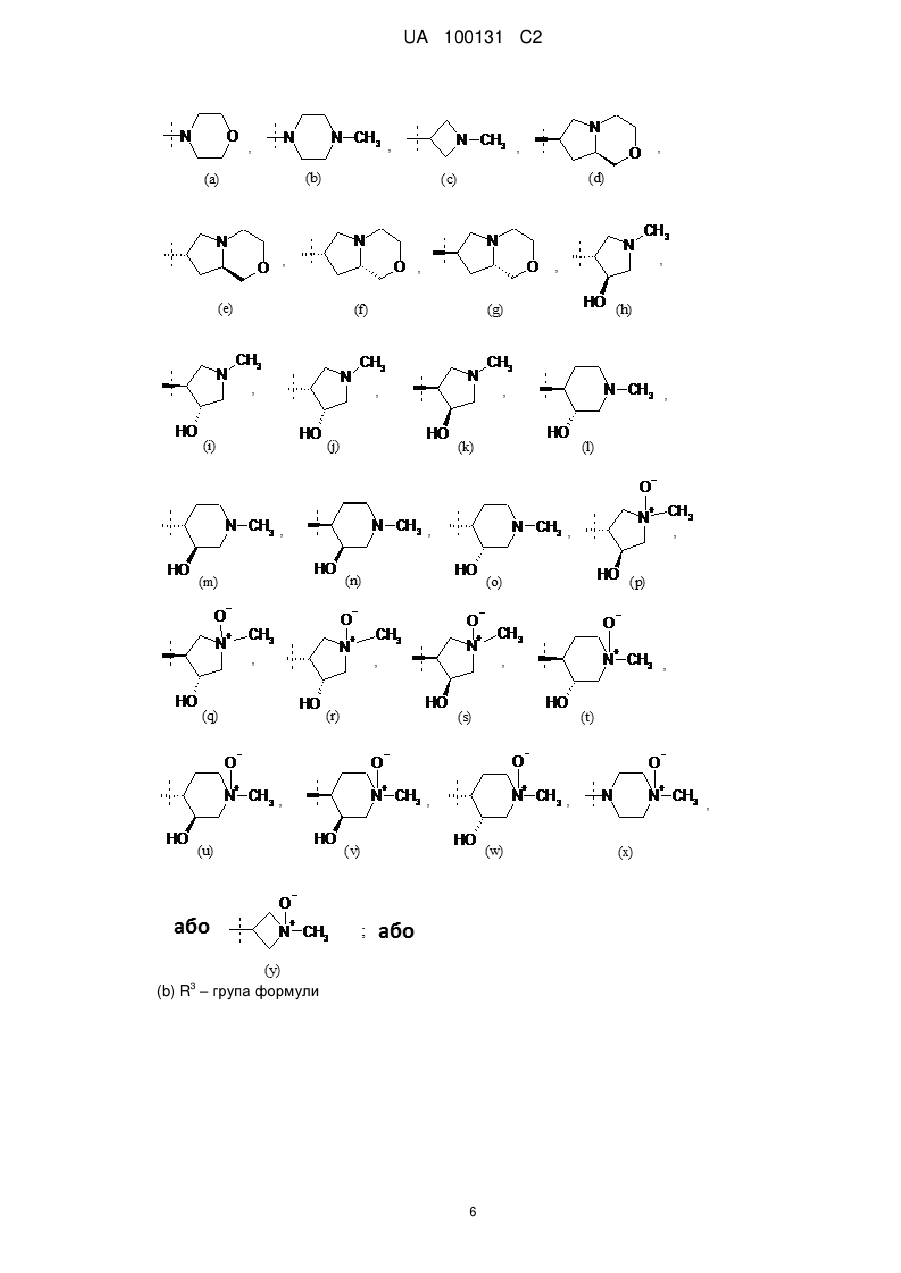
R3 - водень або група формули:
 ,
,  ,
, 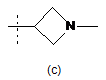 ,
, 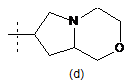 ,
, 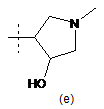 ,
,  ,
, 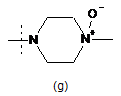 ,
, 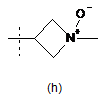 ,
, 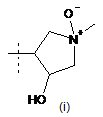 або
або 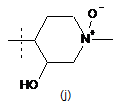 ;
;
та
R4 - група -CN або -C(O)NH2,
або фармацевтично прийнятна сіль такої сполуки.
2. Сполука або сіль за п. 1, де R1 - водень, та R2 - водень або фтор.
3. Сполука або сіль за п. 1, де R1 - водень або фтор, та R2 - водень.
4. Сполука або сіль за п. 1, де кожний з R1 та R2 незалежно від іншого - водень.
5. Сполука або сіль за будь-яким із пп. 1-4, де L - -СН(СН3)-СН2- або безпосередній зв'язок.
6. Сполука або сіль за будь-яким із пп. 1-5, де R3 - група формули:
, , , , , або .
7. Сполука або сіль за будь-яким із пп. 1-6, де R4 - -CN.
8. Сполука або сіль за будь-яким із пп. 1-6, де R4 - -C(O)NH2.
9. Сполука або сіль за п. 1, де
кожний з R1 та R2 незалежно від іншого - водень або фтор;
L - -СН(СН3)-СН2- або безпосередній зв'язок;
R3 - водень або група формули:
, , , , , або ; та
R4 - -CN або -C(O)NH2.
10. Сполука або сіль за будь-яким із пп. 1-9, вибрана з групи, яку складають
(Е)-N-(5-((E)-3-фтор-6Н-дибензо[b,е]оксепін-11-іліденметил)-1-(1-метилазетидин-3-іл)-1,3-дигідробензімідазол-2-іліден)ціанамід;
(E)-N-(5-((E)-(3-фтордибензо[b,е]оксепін-11(6Н)-іліден)метил)-1-((3S,4S)-4-гідрокси-1-метилпіролідин-3-іл)-1Н-бенз[d]імідазол-2(3Н)-іліден)ціанамід;
малеат (E)-N-(5-((E)-(3-фтордибензо[b,е]оксепін-11(6Н)-іліден)метил)-1-((3S,4S)-4-гідрокси-1-метилпіролідин-3-іл)-1Н-бенз[d]імідазол-2(3Н)-іліден)ціанаміду;
(E)-N-[5-((E)-3-фтор-6Н-дибензо[b,е]оксепін-11-іліденметил)-1-((3S,4S)-4-гідрокси-1-метилпіролідин-3-іл)-1,3-дигідробензімідазол-2-іліден]сечовина;
(E)-N-(5-((E)-3-фтор-6Н-дибензо[b,е]оксепін-11-іліденметил)-1-((R)-1-метил-2-морфолін-4-ілетил)-1,3-дигідробензимідазол-2-іліден)сечовина та
(E)-N-(5-((E)-3-фтор-6Н-дибензо[b,е]оксепін-11-іліденметил)-1-((7S,8aR)-гексагідропіроло[2,1-с][1,4]оксазин-7-іл)-1,3-дигідробензімідазол-2-іліден)сечовина.
11. Сполука за п. 10, яка являє собою (E)-N-(5-((E)-(3-фтордибензо[b,е]оксепін-11(6Н)-іліден)метил)-1-((3S,4S)-4-гідрокси-1-метилпіролідин-3-іл)-1Н-бенз[d]імідазол-2(3Н)-іліден)ціанамід або його фармацевтично прийнятну сіль.
12. Сіль за п. 10, яка є малеат (E)-N-(5-((E)-(3-фтордибензо[b,е]оксепін-11(6Н)-іліден)метил)-1-((3S,4S)-4-гідрокси-1-метилпіролідин-3-іл)-1Н-бенз[d]імідазол-2(3Н)-іліден)ціанаміду.
13. Сполука за п. 10, яка являє собою (E)-N-(5-((E)-3-фтор-6Н-дибензо[b,е]оксепін-11-іліденметил)-1-((R)-1-метил-2-морфолін-4-ілетил)-1,3-дигідробензимідазол-2-іліден)сечовину або її фармацевтично прийнятну сіль.
14. Сполука або сіль за будь-яким із пп. 1-13 для застосування у терапії.
15. Сполука або сіль за будь-яким із пп. 1-13 для застосування при лікуванні застійної серцевої недостатності, діабетичної нефропатії, хронічної хвороби нирок, гіпертензії, гіпокаліємії, аритмії міокарда, синдрому Бартера, первинного або вторинного гіперальдостеронізму або синдрому Конна.
16. Сполука або сіль за п. 15 для застосування при лікуванні застійної серцевої недостатності, гіпертензії, діабетичної нефропатії або хронічної хвороби нирок.
17. Фармацевтична композиція, яка містить сполуку або сіль за будь-яким із пп. 1-13 у комбінації з одним або кількома фармацевтично прийнятними носіями, розріджувачами або наповнювачами.
18. Фармацевтична композиція за п. 17, яка містить сполуку, яка являє собою (E)-N-(5-((E)-3-фтор-6Н-дибензо[b,е]оксепін-11-іліденметил)-1-((R)-1-метил-2-морфолін-4-ілетил)-1,3-дигідробензімідазол-2-іліден)сечовину або її фармацевтично прийнятну сіль у комбінації з одним або кількома фармацевтично прийнятними носіями, розріджувачами або наповнювачами.
Текст
Реферат: Цей винахід пропонує сполуку Формули (І): ЯК НЕСТЕРОЇДНІ АНТАГОНІСТИ РЕЦЕПТОРІВ UA 100131 C2 (12) UA 100131 C2 R R O F 2 L R 1 N NH 3 N R 4 Формула (І) та способи лікування фізіологічних розладів, зокрема застійної серцевої недостатності, гіпертензії, діабетичної нефропатії або хронічної хвороби нирок. Цей винахід має відношення до нестероїдних антагоністів рецепторів мінералокортикоїдів. 1 R O 2 R F 3 R L N NH N 4 R UA 100131 C2 5 10 15 20 25 30 35 40 45 50 Цей винахід стосується трициклічних сполук, прийнятних як терапевтичні засоби при лікуванні фізіологічних розладів, сприйнятливих до антагоністів рецепторів мінералокортикоїдів, фармацевтичних композицій, які містять згадані сполуки, способів застосування згаданих сполук для лікування фізіологічних розладів у пацієнтів та проміжних продуктів та способів, прийнятних для синтезу згаданих сполук. Альдостерон, який є головним ендогенним мінералокортикоїдом, регулює гемодинамічний гомеостаз шляхом сприяння реабсорбції натрію та води і виділенню калію після взаємодії з рецептором мінералокортикоїду (MR). У зв'язку з роллю, яку відіграє альдостерон у підтримуванні електролітної та водної рівноваги, антагоністи MR застосовували для лікування численних фізіологічних розладів, в тому числі гіпертензії, гіпокаліємії, аритмій міокарда, синдрому Бартера, а також розладів типу первинного або вторинного гіперальдостеронізму, такого як синдром Конна. Останнім часом антагоністи MR застосовували при лікуванні застійної серцевої недостатності та гострого інфаркту міокарда. Крім того, було доведено, що антагоністи MR є ефективними у передклінічних моделях захворювань нирок та у комбінації зі стандартною терапією для зниження протеїнурії у пацієнтів, які страждають на ниркові розлади, такі як хронічна хвороба нирок, в тому числі на діабетичну нефропатію. Однак відомі антагоністи MR спричиняють супутні ефекти, які обмежують їхню безпечність та/або ефективність. Наприклад, спіронолактон, який є ефективним антагоністом MR, є неселективним та вступає в перехресні реакції з іншими рецепторами ядерних гормонів (наприклад, із рецептором андрогену (AR), рецептором прогестерону (PR) або з рецептором глюкокортикоїду (GR)), які опосередковують інші фізіологічні процеси. Спіронолактонова терапія пов'язана з гіперкаліємією, а також з гінекомастією, еректильною дисфункцією, зниженим лібідо, нерегулярними менструаціями, а також з болями у шлунку. Ерлеренон, який є селективним стосовно до MR у порівнянні з іншими рецепторами ядерних гормонів, також пов'язаний з гіперкаліємією. Отже, в цій галузі постійно існує потреба у способах терапії, пов'язаних із застосуванням альтернатив існуючим антагоністам MR. Метою цього винаходу є запропонувати нестероїдні ліганди MR, які мають активність як антагоністи MR. За варіантом здійснення, якому віддається перевага, метою цього винаходу є запропонувати нестероїдні антагоністи MR, які зв'язуються з MR з підвищеною спорідненістю у порівнянні з AR, PR та GR. За варіантом здійснення, якому віддається більша перевага, метою цього винаходу є запропонувати нестероїдні антагоністи MR, які зв'язуються з MR з підвищеною спорідненістю у порівнянні з AR, PR та GR та мають високу нефро- або кардіопротекторну активність. За варіантом здійснення, якому віддається навіть більша перевага, метою цього винаходу є запропонувати нестероїдні антагоністи MR, які зв'язуються з MR з підвищеною спорідненістю у порівнянні з AR, PR та GR та мають високу нефро- або кардіопротекторну активність, але відрізняються зниженою частотою або ймовірністю спричинення гіперкаліємії. Важливою властивістю будь-якого терапевтичного засобу є здатність цього засобу до спричинення подовження інтервалу QT. Головним механізмом, за допомогою якого терапевтичний агент може спричинити подовження інтервалу QT, є блокування каналів hERG у серцевому м'язі. Блокування каналу hERG заважає проходженню іонів калію через мембрани серцевих клітин, результатом чого є подовження дії клітинних потенціалів, що може спричинити небезпечну серцеву аритмію. Таким чином, за іншим варіантом здійснення цього винаходу, якому віддається перевага, метою винаходу є запропонувати антагоністи MR зі зниженою частотою або ймовірністю блокування каналів hERG. Трициклічні ліганди MR відомі в цій галузі. Наприклад, у WO 04/052847 та WO 05/066161 описані трициклічні стероїдні модулятори рецепторів гормонів, прийнятні при лікуванні розладів, сприйнятливих до модулювання рецепторів мінералокортикоїдів або рецепторів глюкокортикоїдів. Цей винахід грунтується на тому, що певні нові трициклічні сполуки, які відповідають поданій нижче Формулі (I), мають специфічні криві активності, які вказують, що згадані сполуки є прийнятними, що підтверджено випробуваннями in vitro та in vivo, при лікуванні або профілактиці розладів, сприйнятливих до лікування антагоністами рецепторів мінералокортикоїдів. Зокрема, подані як приклади сполуки Формули (I) є ефективними лігандами MR, які протидіють рецептору мінералокортикоїдів. Отже, цей винахід пропонує сполуку Формули (I) 1 UA 100131 C2 1 R O 2 R F 3 R L N NH N 4 R 5 , Формула (I) 1 2 де кожний з R та R незалежно від іншого – водень або фтор; L – -(CH2)2-, -CH(CH3)-CH2- або безпосередній зв'язок; 3 R – водень або група формули: 4 10 15 20 25 R – -CN або -C(O)NH2, або фармацевтично прийнятну сіль такої сполуки. У іншому варіанті здійснення цей винахід пропонує спосіб лікування або профілактики застійної серцевої недостатності, діабетичної нефропатії, хронічної хвороби нирок, гіпертензії, гіпокаліємії, аритмії міокарда, синдрому Бартера, первинного або вторинного гіперальдостеронізму, або синдрому Конна, який включає введення в організм пацієнта, який потребує такого лікування або профілактики, ефективної кількості сполуки Формули (I) або фармацевтично прийнятної солі такої сполуки. У більш конкретному аспекті цей винахід пропонує спосіб лікування або профілактики застійної серцевої недостатності, гіпертензії, діабетичної нефропатії або хронічної хвороби нирок. Крім того, цей винахід пропонує сполуку Формули (I) або фармацевтично прийнятну сіль такої сполуки для застосування при лікуванні або профілактиці застійної серцевої недостатності, діабетичної нефропатії, хронічної хвороби нирок, гіпертензії, гіпокаліємії, аритмії міокарда, синдрому Бартера, первинного або вторинного гіперальдостеронізму, або синдрому Конна. Більш конкретно, цей винахід пропонує сполуку Формули (I) або фармацевтично прийнятну сіль такої сполуки для застосування при лікуванні або профілактиці застійної серцевої недостатності, гіпертензії, діабетичної нефропатії або хронічної хвороби нирок. Крім того, цей винахід пропонує сполуку Формули (I) або фармацевтично прийнятну сіль такої сполуки для застосування у терапії. У іншому варіанті здійснення цей винахід пропонує застосування сполуки Формули (I) або 2 UA 100131 C2 5 10 15 20 25 30 35 40 45 50 55 60 фармацевтично прийнятної солі такої сполуки для виготовлення лікарського засобу для лікування або профілактики застійної серцевої недостатності, діабетичної нефропатії, хронічної хвороби нирок, гіпертензії, гіпокаліємії, аритмії міокарда, синдрому Бартера, первинного або вторинного гіперальдостеронізму, або синдрому Конна. Більш конкретно, цей винахід пропонує застосування сполуки Формули (I) або фармацевтично прийнятної солі такої сполуки для виготовлення лікарського засобу для лікування або профілактики застійної серцевої недостатності, гіпертензії, діабетичної нефропатії або хронічної хвороби нирок. Крім того, цей винахід пропонує фармацевтичну композицію, яка містить сполуку Формули (I) або фармацевтично прийнятну сіль такої сполуки у комбінації з одним або кількома фармацевтично прийнятними носіями, розріджувачами або наповнювачами. Більш конкретно, цей винахід пропонує фармацевтичну композицію для лікування або профілактики застійної серцевої недостатності, гіпертензії, діабетичної нефропатії або хронічної хвороби нирок, яка містить сполуку Формули (I) або фармацевтично прийнятну сіль такої сполуки у комбінації з одним або кількома фармацевтично прийнятними носіями, розріджувачами або наповнювачами. Цей винахід охоплює також нові проміжні продукти та процеси, прийнятні для синтезу сполук Формули (I). Цей винахід стосується також сольватів сполуки Формули (I) або фармацевтично прийнятних солей сполук Формули (I). Таким чином, при вживанні в цьому описі термін "Формула (I)” або назва будь-якої конкретної сполуки Формули (I) охоплює своїм значенням будь-який сольват згаданої сполуки. Приклади фармацевтично прийнятних солей та способи їх одержання добре відомі фахівцям у цій галузі. Дивись, наприклад, Stahl et al., "Handbook of Pharmaceutical Salts: Properties, Selection and Use", VCHA/Wiley-VCH, (2002); Gould P.L., "Salt selection for basic drugs", International Journal of Pharmaceutics, 33: 201-217 (1986); Berge et al., "Pharmaceutical Salts", Journal of Pharmaceutical Sciences, 66, No. 1, (January 1977); та Bastin et al. "Salt Selection and Optimization Procedures for Pharmaceutical New Chemical Entities", Organic Process Research and Development, 4: 427-435 (2000). Особливо слід згадати гідрохлориди, малеїнати та мезилати сполук Формули (I). Сполуки за цим винаходом можуть включати один або кілька хіральних центрів, і томуможуть існувати у різноманітних стереоізомерних конфігураціях. В результаті наявності цих хіральних центрів сполуки за цим винаходом можуть мати форму рацематів, сумішей енантіомерів та індивідуальних енантіомерів, а також діастереомерів та сумішей діастереомерів. За винятком випадків, конкретно вказаних у цьому документі, усі такі рацемати, енантіомери та діастереомери охоплюються обсягом цього винаходу. Енантіомери сполук, які пропонуються цим винаходом, фахівець у цій галузі може розділити, наприклад, застосувавши стандартні способи, такі як описані у J.Jacques, et al., "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, Inc., 1981, а також способи, запропоновані в цьому описі на Схемах, у розділах "Підготовчі синтези" та „Приклади". Конкретні стереоізомери та енантіомери сполук Формули (I) фахівець у цій галузі може одержати, застосовуючи добре відомі способи та процеси, такі як описані у Eliel and Wilen, "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., 1994, Chapter 7; Separation of Stereoisomers, Resolution, Racemization; та Collet and Wilen, "Enantiomers, Racemates, and Resolutions", John Wiley & Sons, Inc., 1981, а також способи, запропоновані в цьому описі на Схемах, у розділах "Підготовчі синтези" та „Приклади". Наприклад, конкретні стереоізомери та енантіомери можна одержати шляхом стереоспецифічних синтезів, у яких застосовуюють енантіомерно та геометрично чисті або енантіомерно чи геометрично збагачені вихідні матеріали. Крім того, конкретні стереоізомери та енантіомери можна розділяти та виділяти такими способами як хроматографія на хіральних стаціонарних фазах, ферментативне розділення або фракційна перекристаллизація солей, одержаних із застосуванням реагентів, які використовують для цієї мети. Позначення "R" та "S" вживаються в цьому описі за звичайними правилами органічної хімії для позначення конкретних конфігурацій хіральних центрів. Позначення “()”, "R/S" або "RS" стосуються рацемічної конфігурації хіральних центрів. Частковий перелік пріоритетів та розгляд питань стереохімії міститься у "Nomenclature of Organic Compounds: Principles and Practice" (J.H. Fletcher, et al., eds., 1974). Як зрозуміло для фахівця у цій галузі, молекули, які містять подвійний зв'язок вуглець-вуглець або вуглець-азот, можуть існувати у формі геометричних ізомерів. Для позначення таких конкретних ізомерів звичайно застосовуються дві системи: "цистранс" та "E-Z", залежно від того, чи є групи, приєднані до кожного з атомів, з'єднаних подвійним зв'язком, однаковими або різними. Розгляд питань геометричної ізомерії та найменування конкретних ізомерів можна знайти у March, "Advanced Organic Chemistry", John Wiley & Sons, 3 UA 100131 C2 5 10 15 20 25 30 35 40 45 50 55 60 1992, Chapter 4. Усі такі геометричні ізомери, а також суміші індивідуальних ізомерів, охоплюються та пропонуються цим винаходом. Сполуки за цим винаходом можуть входити до складу фармацевтичної композиції як її компоненти. Таким чином, фармацевтична композиція, яка містить сполуку Формули (I) або фармацевтично прийнятну сіль такої сполуки у комбінації із фармацевтично прийнятним носієм, розріджувачем або наповнювачем, є важливим варіантом здійснення цього винаходу. Приклади фармацевтичних композицій та способи їх виготовлення є добре відомими в цій галузі. Дивись, наприклад, REMINGTON: THE SCIENCE AND PRACTICE OF PHARMACY (A. Gennaro, et al., th eds., 19 ed., Mack Publishing (1995)). До ілюстративних композицій, які містять сполуки Формули (I), належать, наприклад, такі: сполука Формули (I) у суспензії з 0,5 % карбоксиметилцелюлози, 0,25 % Полісорбату 80 та 2,7 % NaCl; або сполука Формули I у суспензії з 1 % карбоксиметилцелюлози та 0,25 % Полісорбату 80; або сполука Формули I у суспензії з 1 % карбоксиметилцелюлози, 0,25 % Полісорбату 80 та 0,05 % протиспінювального засобу AntiFoam 1510 в очищеній воді. Однак слід мати на увазі, що композиція за цим винаходом, якій віддається перевага, містить сполуку Формули (I) або фармацевтично прийнятну сіль такої сполуки у складі капсули або таблетки. Сполука Формули (I) або композиція, яка містить сполуку Формули (I), може бути введена в організм будь-яким шляхом, який забезпечує біодоступність сполуки, в тому числі пероральним та парентеральним шляхами. Для фахівця у цій галузі зрозуміло, що розмір частинок фармацевтичного засобу може впливати на його розчинність in vivo, що, у свою чергу, може впливати на засвоєння засобу. Термін "розмір частинок" у значенні, вжитому в цьому описі, стосується діаметра частинки фармацевтичного засобу, визначеного звичайними способами, такими як розсіяння лазерного випромінення, дифракція лазерного випромінення, розсіяння Мі (розсіяння світла на кульових частинках), фракціональна седиментація, спектроскопія кореляції фотонів тощо. Якщо фармацевтичні засоби мають низьку розчинність, то незначний або зменшений розмір частинок може сприяти розчиненню і таким чином підвищити абсорбування засобу. Дивись Amidon et al., Pharm. Research, 12; 413-420 (1995). Способи зменшення або регулювання розмірів частинок широко відомі, і до них належать розмелювання, вологе подрібнення, тонке подрібнення тощо. Інший спосіб регулювання розміру частинок включає одержання фармацевтичного засобу у формі наносуспензії. Конкретний варіант здійснення цього винаходу включає сполуку Формули (I) або фармацевтичну композицію, яка містить сполуку Формули (I), причому згадана сполука має розмір d90 частинок (тобто гранулометричний склад, в якому 90 % частинок мають розмір, який дорівнює вказаному значенню або є меншим ніж вказане значення) менше ніж приблизно 50 мкм. Більш конкретний варіант включає сполуку Формули I, яка має розмір d90 частинок менше ніж приблизно 10 мкм. У значенні, вжитому в цьому описі, термін "пацієнт" стосується людини або іншого ссавця, такого як собака, кіт, корова, мавпа, кінь або вівця. Більш конкретно, термін "пацієнт" стосується людини. Термін "лікування" (або "лікувати") у значенні, вжитому в цьому описі, охоплює запобігання, попередження, обмеження, уповільнення, припинення або обернення напрямку розвитку або тяжкості існуючого симптому або розладу. Термін "попередження" (або "попереджувати", або "профілактика") у значенні, вжитому в цьому описі, означає виключення, обмеження або інгібування виникнення або наявності симптому або розладу. Як відомо фахівцям у цій галузі, фізіологічні розлади можуть мати форму "хронічних" станів або "гострих" нападів. Таким чином, лікування розладів охоплює як гострі випадки, так і хронічні стани. При гострому нападі сполуку вводять на початку виникнення симптомів і припиняють вводити при зникненні симптомів, в той час як при хронічних станах лікування продовжують протягом усього періоду захворювання. У значенні, вжитому в цьому описі, термін "ефективна кількість" означає кількість або дозу сполуки Формули (I), яка при введенні разовою дозою або кількома дозами в організм пацієнта забезпечує бажаний вплив на пацієнта, якого піддають діагностиці або лікуванню. Ефективна кількість може бути легко визначена лікарем-куратором як фахівцем у цій галузі, з урахуванням численних чинників, таких як вид ссавця, розміри його тіла, вік та загальний стан здоров'я; конкретне захворювання, яке підлягає лікуванню; ступінь тяжкості захворювання; індивідуальна реакція пацієнта; конкретна застосовувана сполука; режим застосування; властивості біодоступності застосовуваного препарату; обраний режим дозування; та застосування будьяких супутніх лікарських засобів. При використанні згідно зі способами та варіантами застосування за цим винаходом сполуки та композиції за цим винаходом можуть бути введені або окремо, або в комбінації з відомими терапевтичними засобами, які застосовують для лікування конкретного розладу або стану. 4 UA 100131 C2 5 10 15 20 25 Якщо сполуки та композиції за цим винаходом застосовують як частину комбінації, сполуку Формули (I) або композицію, яка містить таку сполуку, можна застосовувати окремо або як складову частину лікарської форми, яка містить терапевтичний засіб, з яким слід комбінувати згадану сполуку або композицію. Вжите у цьому описі позначення “ “ означає зв'язок, який виступає над площиною сторінки, а позначення “ “ означає зв'язок, заглиблений під площину сторінки. Конкретні аспекти цього винаходу Подані нижче переліки включають кілька груп конкретних конфігурацій, замісників та змінних варіантів сполук Формули (I). Мається на увазі, що сполуки Формули (I), які мають такі конкретні конфігурації, замісники та змінні варіанти, а також способи, застосування та композиції, які містять такі сполуки, відповідають конкретним аспектам цього винаходу. Таким чином, за одним конкретним аспектом цього винаходу сполука Формули (I) являє 3 4 собою сполуку, в якій L, R та R мають будь-яке зі значень, визначених у цьому описі, та: 1 2 (a) R – водень, та R – водень або фтор; або 1 2 (b) R – фтор, та R – водень або фтор; або 1 2 (e) R – водень або фтор, та R – водень; або 1 2 (d) R – водень або фтор, та R – фтор; або 1 2 (e) кожний з R та R незалежно від іншого – водень. За іншими конкретними конкретними аспектами цього винаходу сполука Формули (I) являє 1 2 3 4 собою сполуку, в якій R , R , R та R мають будь-яке зі значень, визначених у цьому описі, та: (a) L – -CH(CH3)-CH2- або безпосередній зв'язок; або (b) L – -CH(CH3)-CH2-; або (c) L – безпосередній зв'язок. За іншими конкретними аспектами цього винаходу сполука Формули (I) являє собою 1 2 4 сполуку, в якій R , R , L та R мають будь-яке зі значень, визначених у цьому описі, та: За переліченими нижче конкретними аспектами цього винаходу сполука Формули (I) являє 1 2 4 собою сполуку, в якій R , R , L та R мають будь-яке зі значень, визначених у цьому описі, та: 3 (a) R – водень або група формули 5 UA 100131 C2 3 (b) R – група формули 6 UA 100131 C2 3 (b) R – група формули 7 UA 100131 C2 5 За іншими конкретними аспектами цього винаходу сполука Формули (I) являє собою 1 2 3 сполуку, в якій R , R , L та R мають будь-яке зі значень, визначених у цьому описі, та: 4 (a) R – -CN, або 4 (b) R – -C(O)NH2. За більш конкретним аспектом цього винаходу сполука Формули (I) являє собою сполуку, в 1 2 якій кожний з R та R незалежно від іншого – водень або фтор; L – -CH(CH3)-CH2- або безпосередній зв'язок; 3 R – водень або група формули: 10 8 UA 100131 C2 4 5 R – -CN або -C(O)NH2, або фармацевтично прийнятну сіль такої сполуки. За ще більш конкретним аспектом цього винаходу сполука Формули (I) являє собою сполуку, 1 2 в якій кожний з R та R незалежно від іншого – водень або фтор; L – -CH(CH3)-CH2- або безпосередній зв'язок, за умови, що якщо L – 3 -CH(CH3)-CH2-, то R – група формули: N O , 3 а якщо L – безпосередній зв'язок, то R – група формули: 10 15 4 R – -CN або -C(O)NH2, або фармацевтично прийнятну сіль такої сполуки. За іншими конкретними аспектами цього винаходу пропонуються сполуки поданих нижче Формули I(a) та Формули I(b). Мається на увазі, що сполуки Формули I(a) та Формули I(b), а також способи та варіанти застосування таких сполук є іншими конкретними аспектами цього винаходу. Таким чином, за одним конкретним аспектом цього винаходу пропонується сполука Формули I(a): 1 R O 2 R F 3 R L N NH N CN , Формула I(a) 9 UA 100131 C2 1 2 де кожний з R та R незалежно від іншого – водень або фтор; L – -CH(CH3)-CH2- або безпосередній зв'язок; та 3 R – водень або група формули: 5 або фармацевтично прийнятна сіль такої сполуки. За ще більш конкретним аспектом цього винаходу пропонується сполука Формули I(a), в якій 1 2 кожний з R та R незалежно від іншого – водень або фтор; та L – -CH(CH3)-CH2- або безпосередній зв'язок, за умови, що якщо L – 3 -CH(CH3)-CH2-, то R – група формули: N 10 O , 3 а якщо L – безпосередній зв'язок, то R – група формули: або фармацевтично прийнятна сіль такої сполуки. За одним конкретним аспектом цього винаходу пропонується сполука Формули I(b): 10 UA 100131 C2 1 R O 2 R F 3 R L N NH N NH2 O 5 10 , Формула I(b) 1 2 де кожний з R та R незалежно від іншого – водень або фтор; L – -CH(CH3)-CH2- або безпосередній зв'язок; та 3 R – група формули: або фармацевтично прийнятна сіль такої сполуки. За ще більш конкретним аспектом цього винаходу пропонується сполука Формули I(b), в якій 1 2 кожний з R та R незалежно від іншого – водень або фтор; та 3 L – -CH(CH3)-CH2- або безпосередній зв'язок, за умови, що якщо L – -CH(CH3)-CH2-, то R – група формули: N O , 3 а якщо L – безпосередній зв'язок, то R – група формули: 15 20 25 30 або фармацевтично прийнятна сіль такої сполуки. Крім того, за найбільш конкретними аспектами цього винаходу пропонуються сполуки Формули (I), подані в цьому описі як приклади, найконкретніше (E)-N-(5-((E)-3-фтор-6H-дибензо[b, e]оксепін-11-іліденметил)-1-(1-метилазетидин-3-іл)-1,3дигідробензимідазол-2-іліден)ціанамід; (E)-N-(5-((E)-(3-фтордибензо[b, e]оксепін-11(6H)-іліден)метил)-1-((3S, 4S)-4-гідрокси-1метилпіролідин-3-іл)-1H-бенз[d]імідазол-2(3H)-іліден)ціанамід; малеїнат (E)-N-(5-((E)-(3-фтордибензо[b, e]оксепін-11(6H)-іліден)метил)-1-((3S, 4S)-4гідрокси-1-метилпіролідин-3-іл)-1H-бенз[d]імідазол-2(3H)-іліден)ціанаміду; (E)-N-[5-((E)-3-фтор-6H-дибензо[b, e]оксепін-11-іліденметил)-1-((3S, 4S)-4-гідрокси-1метилпіролідин-3-іл)-1,3-дигідробензимідазол-2-іліден]сечовина; (E)-N-(5-((E)-3-фтор-6H-дибензо[b, e]оксепін-11-іліденметил)-1-((R)-1-метил-2-морфолін-4-ілетил)-1,3-дигідробензимідазол-2-іліден)сечовина або (E)-N-(5-((E)-3-фтор-6H-дибензо[b, e]оксепін-11-іліденметил)-1-((7S, 8aR)-гексагідропіроло[2,1-c][1,4]оксазин-7-іл)-1,3-дигідробензимідазол-2-іліден)сечовина. Сполуки Формули (I) можна одержати за різноманітними методиками, відомими у цій галузі, а також за методиками, представленими нижче на Схемах, у розділах "Підготовчі синтези" та "Приклади". Однак подані нижче описи не слід розглядати як такі, що обмежують будь-яким чином обсяг цього винаходу. Наприклад, конкретні стадії синтезу в межах кожного з описаних шляхів можна комбінувати різними способами або поєднувати стадії з різних схем для 11 UA 100131 C2 5 10 15 одержання інших сполук Формули (I). Продукти, одержувані на кожній стадії поданих нижче схем, можна виділяти звичайними способами, в тому числі екстрагуванням, випарюванням, осадженням, хроматографією, фільтруванням, розтиранням, кристалізацією тощо. Замісники, якщо не вказано інакше, відповідають поданим вищевизначенням. Реагенти та вихідні матеріали легко доступні для фахівців у цій галузі. Інші необхідні реагенти та вихідні матеріали можуть бути виготовлені за методиками, вибраними з сукупності стандартних методик органічної хімії та хімії гетероциклічних сполук, способами, аналогічними способам синтезу відомих сполук аналогічної структури та за методиками, описаними нижче в розділах "Підготовчі синтези" та „Приклади", в тому числі за будь-якими новими методиками. Подані нижче абревіатури, вжиті в цьому описі, мають такі значення: "MeOH" означає метанол; "EtOH" означає етанол; "EtOAc" означає етилацетат; "DMF" означає диметилформамід; "DMSO" означає диметилсульфоксид; "TFA" означає трифтороцтову кислоту; "THF" означає тетрагідрофуран; "DEAD" означає діетилазодикарбоксилат; "DIAD" означає діізопропілдіазодикарбоксилат; "NBS" означає N-бромсукцинімід; "MCPBA" означає мхлорпероксибензойну кислоту; "tBOC" або "boc" означає трет-бутоксикарбоніл; "rac" означає рацемічний; "prep" означає підготовчий синтез; "ex" означає приклад; та "HPLC" означає рідинну хроматографію високої ефективності. 20 Проміжний продукт формули (3) можна одержати відповідно до реакцій, показаних на Схемі 1. 25 30 35 40 Згідно зі Схемою 1 амін формули (1) вводять в реакцію з 4-бром-1-фтор-2-нітробензолом за схемою нуклеофільного ароматичного заміщення (SNAr) для одержання амінонітрофенілброміду формули (3). Наприклад, амін (1) можна ввести в реакцію в інертному розчиннику, такому як етилацетат, THF, або у протоновмісному розчиннику, такому як етанол, при температурі в межах від кімнатної до температури кипіння розчинника. Для фахівця у цій галузі зрозуміло, що сполуки формули (1) можна легко одержати способами, аналогічними описаним у цьому документі, або із застосуванням методик, поширених у цій галузі. Наприклад, метилзаміщений або незаміщений морфолінілетиламін можна одержати шляхом мезилювання N-трет-бутилкарбоксилетаноламіну з подальшою заміною мезилату на морфолін з одержанням бажаного морфолінілетиламіну. Слід також зазначити, що вибір та застосування прийнятних груп захисту добре відомі та поширені в цій галузі (дивись, наприклад, „Protecting Groups in Organic Synthesis, Theodora Greene (Wiley-Interscience)). Від амінної функціональної групи, такої як присутня в радикалі азетидинілу, можна відщепити групу захисту, і потім ввести її в наступну реакцію для одержання додаткових сполук за цим винаходом. Наприклад, після відщеплення групи захисту від третбутилового складного ефіру 3-(4-бром-2-нітрофеніламіно)азетидин-1-карбонової кислоти в кислотному середовищі азетидин можна алкілувати способом відновлювального амінування, наприклад, із формальдегідом, для одержання як проміжного продукту N-метилазетидинілу. 12 UA 100131 C2 Проміжний продукт формули (9) можна одержати відповідно до реакцій, показаних на Схемі 2. 5 10 15 20 25 30 На Стадії 1 Схеми 2 складний ефір формули (4) відновлюють до спирту формули (5). Для фахівця у цій галузі буде зрозуміло, що існують численні способи відновлення складних ефірів. Наприклад, складний ефір формули (4) обробляють боргідридом літію при температурі від приблизно 0C до 40C в інертному розчиннику, такому як діетиловий ефір. Після гасіння реакції через 10 хв можна виділити азид спирту формули (5), який потім відновлюють до аміну на Стадії 2. Для одержання аміноспирту формули (6) можна виконати відновлення азиду із застосованням відомих в цій галузі умов каталітичного гідрування. На Стадії 3 Схеми 2 4-бром-1-фтор-2-нітробензол піддають реакції SNAr з аміном формули (6), як описано вище для Схеми 1. На Стадії 4 від феніламінопіролідину формули (7) відщеплюють групу захисту, застосовуючи кислотне середовище, таке як хлороводень або трифтороцтова кислота, і виконують реакцію замикання циклу на Стадії 5, одержуючи оксезинон формули (8). Замикання циклу досягається застосуванням хлорацетилхлориду у присутності неорганічної основи, такої як гідроксид натрію, при підтриманні pH=10-12. Реакцію проводять у суміші THF з водою (приблизно 1:1). На Стадії 6 Схеми 2 оксезинон формули (8) відновлюють до оксезину формули (9). Карбонільну функціональну групу відновлюють, застосовуючи комплекс борану з тетрагідрофураном або комплекс борану з диметилсульфідом в інертному розчиннику, такому як THF, при температурі від приблизно 0C до температури кипіння розчинника. Хіральні піролідини формули (4) відомі в цій галузі, і їх можна легко одержати зі стереоізомерів 3-гідроксипроліну. Спирт можна мезилювати, а потім замістити з утворенням азиду з оберненням стереохімічної конфігурації. За альтернативним варіантом спирт можна перетворити у бромід в умовах реакції Міцунобу, застосовуючи тетрабромід вуглецю, з оберненням стереохімічної конфігурації, з подальшим заміщенням іоном азиду для повторного обернення стереохімічної конфігурації. Транс-гідроксипролін можна перетворити у цисконфігурацію шляхом оброблення оцтовою кислотою та оцтовим ангідридом при 90C (C.G. Levins and C.E. Schafmeister, J. Org. Chem., 2005, 70, 9002-9008). 13 UA 100131 C2 5 10 15 20 25 Проміжний продукт формули (14a або 14b) або формули (17a або 17b) можна одержати відповідно до реакцій, показаних на Схемі 3. На Стадії 1a Схеми 3 рацемічний транс-гідроксибензиламінпіролідин формули (10) можна розділити на ізомери, застосовуючи відповідну енантіомерно чисту кислоту. Наприклад, обробка (+)-мигдалевою кислотою у прийнятному розчиннику, такому як ацетонітрил із приблизно 1 % води, забезпечує селективну перекристалізацію (S, S)-гідроксибензиламін (+)манделату з дуже високим енантіомерним надлишком. Цю сіль можна потім нейтралізувати, застосовуючи відповідну основу, таку як водний розчин карбонату калію. Для одержання очищеного (R, R)-енантіомеру на Стадії 1b маточний розчин після вищезгаданої перекристалізації, збагачений тепер (R, R)-енантіомером, можна випарити, і обробити (-)мигдалевою кислотою у розчиннику, такому як ацетонітрил, який містить приблизно 2,5 % води. На Стадії 2 шляхом оброблення вільного (S, S)- або (R, R)-гідроксибензиламіну у стандартних умовах гідрування із застосуванням відомих каталізаторів, таких як паладій на вугіллі, у звичайно застосовуваному розчиннику, такому як метанол, н-бутанол або тетрагідрофуран, одержують відповідний (S, S)- або (R, R)-гідроксиламін. На Стадії 3 Схеми 3 шляхом проведення реакції SNAr одержаного амінопіролідину формули (12a або 12b) з 4-бром-1-фтор-2-нітробензолом одержують феніламінопіролідин формули (13a або 13b). Цю реакцію можна виконувати у присутності відповідної основи, такої як триетиламін, у звичайному розчиннику, такому як тетрагідрофуран або етилацетат. На Стадії 4 Схеми 3 хіральну конфігурацію гідроксильної групи піролідину формули (13a або 13b) перетворюють з (S, S) та (R, R) відповідно у (3R, 4S) та (3S, 4R). Хіральне перетворення здійснюють шляхом проведення реакції Міцунобу. Для фахівця у цій галузі зрозуміло, що в техніці використовують різноманітні умови реакції Міцунобу. Наприклад, спирт формули (15a або 15b) розчиняють у відповідному безводному розчиннику, такому як THF, CH2Cl2, толуол тощо, і обробляють триалкіл- або триарилфосфіном, таким як Me3P, Bu3P або Ph3P, та діалкілазодикарбоксилатом, таким як DEAD або DIAD. 14 UA 100131 C2 5 10 15 20 25 30 На Стадії 5 3S, 4R- та 4S, 3R-хлорацетоксіефір формули (15a або 15b) гідролізують з одержанням гідроксипіролідину формули (16a або 16b). Гідроліз виконують із використанням неорганічної основи, такої як водний розчин гідроксиду літію, у розчиннику, такому як метанол, при температурі від 0C до 50C протягом 4-24 год. На Стадії 6 Схеми 3 від чотирьох діастереомерних амінопіролідинів формули (13a або 13b) та формули (16a або 16b) відщеплюють групи захисту, застосовуючи кислотне середовище. Відщеплення трет-бутоксикарбонільних груп захисту добре відоме в цій галузі. Перевага віддається проведенню реакції в інертному розчиннику, такому як етилацетат, оброблений безводним хлороводнем при температурі приблизно 0C протягом 10 хв. Одержаний NHпіролідин піддають відновлювальному амінуванню, і одержують N-метилпіролідин формули (14a або 14b) та формули (17a або 17b). Реакцію проводять в інертному розчиннику, такому як дихлорметан, THF, або за варіантом, якому віддається перевага, ацетонітрил, із застосуванням водного розчину формальдегіду. Реакційну суміш обробляють відновлювачем, таким як боргідрид натрію, ціаноборгідрид натрію, або за варіантом, якому віддається перевага, триацетоксиборгідрид натрію, при температурі від приблизно 0C до 40C протягом 4-24 год. Для фахівця у цій галузі зрозуміло, що сполуки формули (10) можна легко одержати способами, аналогічними описаним у цьому документі, або за допомогою прийнятих у цій галузі методик. Наприклад, до 3-піроліну можна приєднати захисну групу шляхом оброблення ди-третбутилдикарбонатом, після чого бромувати у суміші DMSO/вода з одержанням транс-3-бром-4гідроксипіролідину (A. Kamal et. al. Tetrahedron Lett., 2004, 45, 8057-8059; A. Kamal et. al. Tetrahedron Asymmetry, 2006, 17, 2876-2883). При реакції з гідроксидом натрію утворюється in situ епоксид, який вводять у реакцію з бензиламіном, і одержують рацемічний трансгідроксибензиламінпіролідин формули (10). За більш безпосереднім способом епоксид можна прямо одержати та виділити шляхом окиснення 3-піроліну MCPBA. За ще одним альтернативним варіантом синтезу 2,4-дихлор-2-бутен можна ввести в реакцію з третбутилкарбаматом, і одержати 1-Boc-3-піролін (Y. Tsuzuki, et. al. Tetrahedron Asymmetry, 2001, 12, 2989-2997). Можна взагалі виключити застосування групи захисту трет-Boc шляхом використання для реакції метиламіну замість трет-бутилкарбамату. Подальше окиснення до епоксиду з використанням MCPBA та розкриття циклу з використанням аміаку дає рацемічний транс-4-аміно-1-метилпіролідин-3-ол. Цю сполуку можна ввести в реакцію з 1-бром-4-фтор-3нітробензолом, і одержати сполуку формули (14a або 14b) у вигляді суміші енантіомерів. Розділення цих енантіомерів можна виконати відомими у цій галузі способами, такими як хіральна HPLC або перекристалізація солі з енантіомерно чистою карбоновою кислотою, такою як D-(-) або L-(+)-винна кислота тощо. 35 15 UA 100131 C2 5 10 15 20 25 30 На Стадії 1 Схеми 4 транс-аміногідроксипіперидин формули (18) піддають відновлювальному амінуванню з бензальдегідом, і одержують бензиламінопіперидин формули (19). Для фахівця у цій галузі зрозуміло, що існують різноманітні умови, прийнятні для виконання відновлювального амінування. Наприклад, реакція проходить у розчиннику, такому як етанол, у присутності оцтової кислоти та відновлювача, такого як ціаноборгідрид натрію. На Стадії 2a Схеми 4 рацемічний транс-гідроксибензиламінопіперидин формули (19) розділяють, застосовуючи відповідну енантіомерно чисту кислоту. Наприклад, оброблення (+)винною кислотою у відповідному розчиннику, такому як ацетонітрил із домішкою приблизно 5 % води, забезпечує селективну перекристалізацію (3R, 4R)-гідроксибензиламін (+)-тартрату формули (20a) з дуже високим діастереомерним надлишком. Аналогічно, на Стадії 2b рацемічний матеріал перекристалізують з (-)-винною кислотою у розчиннику, такому як ацетонітрил з домішкою приблизно 2,5 % води, та одержують очищений (S, S)-енантіомер формули (20b). На Стадії 3 від розділених 3R, 4R- та 3S, 4S-енантіомерів формули (20a) та (20b), відповідно, можна відщепити бензильну групу шляхом каталітичного гідрування, і одержати аміногідроксипіперидин формули (21a або 21b). Спочатку тартрат промивають розчином карбонату натрію або калію з подальшою екстракцією дихлорметаном для видалення винної кислоти. Вільний бензиламін потім гідрують в інертному розчиннику, такому як етанол або метанол, із застосуванням 5 % або 10 % паладію на вугіллі. На Стадії 4 Схеми 4 аміногідроксипіперидин формули (21a або 21b) перетворюють у феніламінопіперидин формули (22a або 22b), як показано на Стадії 3 Схеми 3. На Стадії 5 від феніламінопіперидину формули (22a або 22b) відщеплюють групу захисту та алкілують шляхом відновлювального амінування, одержуючи N-метилпіперидин формули (23a або 23b). Реакція проходить у суміші оцтової кислоти з водою у присутності формальдегіду та відновлювача, такого як ціаноборгідрид натрію.4-аміно-3-гідроксипіперидини формули (18) відомі в цій галузі, і їх можна легко одержати з 4-піперидону шляхом відновлення до 4гідроксипіперидину, при захисті boc або іншою групою захисту. Спирт можна мезилувати, перетворити в олефін, після чого епоксидувати, одержуючи рацемічний трет-бутиловий складний ефір цис-7-окса-3-азабіцикло[4.1.0]гептан-3-карбонової кислоти. Епоксидний цикл потім розкривають, застосовуючи аніон азиду, для здійснення транс-регіохімічного перетворення, після чого відновлюють для одержання аміногідроксипіперидину формули (18). 16 UA 100131 C2 5 10 15 20 25 30 Сполуку формули (30) або формули (31) можна одержати відповідно до реакцій, показаних 1 2 на Схемі 5. Прийнятною сполукою Формули (24) є сполука, де R та R відповідають визначенням, поданим для Формули (I), а X=Br або I. Прийнятними сполуками формули (30) або 1 2 3 формули (31) є сполуки, де R , R , L та R відповідають визначенням, поданим для Формули (I). На Стадії 1 та Стадії 2 Схеми 5 галогенбензилфеніловий простий ефір формули (24) піддають подвійному сполученню за Геком (Heck) з метил- або етилакрилатом, і одержують вініловий складний ефір формули (26) з регіохімічною конфігурацією E при подвійному зв'язку. Реакція відбувається у присутності паладієвого каталізатора, такого як тетракіс(трифенілфосфін)паладій (0), хлорид паладію або, відповідно до варіанта, якому віддається перевага, ацетат паладію (II). Реакційна суміш містить основу, таку як триетиламін, карбонат калію, ацетат натрію або четвертинна амонієва сіль, така як бромід тетра-нбутиламонію. Реакція проходить в інертному розчиннику, такому як DMF або N-метилпіролідин, при температурі від приблизно 40C до 130C протягом 4-48 год. з утворенням проміжного продукту формули (25). Цей продукт виділяють, після чого циклізують з одержанням вінілового складного ефіру формули (26) у приблизно тих самих умовах реакції, але при вищій температурі, наприклад, від 90C до 150C. За альтернативним варіантом галогенбензилфеніловий простий ефір формули (24) можна перетворити безпосередньо у вініловий складний ефір формули (26) в одному реакторі, без виділення проміжного продукту формули (25), при температурі від приблизно 90C до 150C. На Стадії 3 складний ефір формули (26) гідролізують, і перетворюють отриману речовину у вінілбромід формули (27). Гідроліз можна виконувати із застосуванням неорганічної основи, такої як гідроксид калію, натрію або, за варіантом, якому віддається перевага, гідроксид літію. Одержану карбонову кислоту потім можна обробити оцтовою кислотою та NBS при температурі від 40C до 100C. На Стадії 4 Схеми 5 вінілбромід формули (27) перетворюють у вінілпінаколборонат формули (28). Вінілбромід вводять у реакцію з біс(пінаколат)дибором у присутності основи, такої як ацетат цезію або калію, в інертному розчиннику, такому як THF, 1,2-диметоксіетан, або, за варіантом, якому віддається перевага, 1,4-діоксан. Реакцію проводять при температурі від приблизно 40C до температури кипіння розчинника, у присутності паладієвого каталізатора, такого як дихлорид 1,1’-біс(дифенілфосфін)фероценпаладію (II) (Pd2(dppf)Cl2) або, за 17 UA 100131 C2 5 10 15 20 25 30 35 40 45 50 55 60 альтернативним варіантом, трис(дибензиліденацетон)дипаладію (0) (Pd2(dba)3) у присутності фосфінового ліганду, такого як трициклогексилфосфін. На Стадії 5 вінілпінаколборонат формули (28) вводять в реакцію з амінонітробромбензолом в умовах перехресного сполучення за Судзукі (Suzuki), і одержують вінілнітрофенільний проміжний продукт формули (29). Для фахівця у цій галузі буде зрозуміло, що існують різноманітні умови, прийнятні для полегшення протікання таких реакцій перехресного сполучення. Умови реакції включають застосування прийнятного розчинника, такого як суміш діоксан/вода або суміш THF/метанол. Реакцію виконують у присутності основи, такої як карбонат натрію, метилат натрію, карбонат калію або ацетат калію. Реакція відбувається у присутності паладієвого каталізатора, такого як тетракістрифенілфосфін-паладій (0), в інертній атмосфері при температурі від приблизно 70C до 120C протягом часу, який становить від приблизно 8 год. до 24 год. На Стадії 6 та Стадії 7 Схеми 5 вінілнітрофенільний проміжний продукт формули (29) відновлюють до діамінофенільного проміжного продукту (не показаного на схемі) у стандартних умовах гідрування з 5 % або 10 % платини на вугіллі або з 5 % або 10 % паладію на вугіллі у відповідному розчиннику, такому як тетрагідрофуран, етилацетат або ізопропанол. Після видалення каталізатора діамінофенільний проміжний продукт можна перетворити у ціаногуанідин формули (30), використовуючи дифеніл-N-ціанокарбонімідат, при температурі від кімнатної до 100C. Застосовують інертний розчинник, такий як ізопропанол, піридин або THF, у присутності або при відсутності неорганічної основи, такої як бікарбонат натрію. Очищення кінцевого продукту можна здійснювати звичайними для фахівців у цій галузі способами, такими як хроматографія або перекристалізація. На Стадії 8 Схеми 5 ціаногуанідин формули (30) гідролізують до сечовини формули (31) у кислотному середовищі. Реакція проходить у 4 н хлористоводневій кислоті у суміші діоксан/вода або TFA/вода протягом часу, який становить від 4 год. до 72 год., при температурі від кімнатної до приблизно 80C. Для фахівця у цій галузі зрозуміло, що сполуки формули (24) можна легко одержати способами, аналогічними описаним у цьому документі, або за допомогою методик, відомих у цій галузі. Наприклад, 2-бром-5-фторфенол або 2-бром-5-хлорфенол алкілують 2-йод- або 2бромбензилбромідом, одержуючи феніловий простий ефір формули (24). При одержанні сполук 1 2 формули (24), де R або R =F, необхідний бензилбромід одержують, наприклад, шляхом оброблення літієм 1-бром-3-фторбензолу та проведення реакції з N-метил-N-фенілформамідом для одержання альдегіду. Подальше відновлення до бензилового спирту та проведення реакції з бромувальним засобом, таким як трибромід фосфору, забезпечує одержання 1-бром-3фторбензилброміду. Визначення біологічної активності У цьому описі позначення "Kd" означає рівноважну константу дисоціації для комплексу ліганд-рецептор; "Ki" означає рівноважну константу дисоціації для комплексу лікарський засібрецептор і вказує на таку концентрацію лікарського засобу, яка у стані рівноваги буде зв'язувати половину центрів зв'язування; "Kb" означає рівноважну константу дисоціації для комплексу антагоніст-рецептор; "IC50" означає концентрацію лікарського засобу, яка відповідає 50 % максимальної інгібувальної реакції, можливої для цього засобу, або, за альтернативним варіантом, концентрації лікарського засобу, яка відповідає 50 % заміщення ліганду, який зв'язується з рецептором; "EC50" означає концентрацію лікарського засобу, яка відповідає 50 % максимальної реакції, можливої для цього засобу; і "ED50" означає дозу введеного лікарського засобу, яка забезпечує 50 % максимальної реакції на цей засіб. Випробування зв'язування ядерних рецепторів стероїдних гормонів Для випробувань конкурентного зв'язування рецептор-ліганд для визначення Ki застосовують лізати людських ембріональних ниркових клітин HEK293 з надлишковою експресією людського MR (рецептора мінералокортикоїду), GR (рецептора глюкокортикоїду), AR (рецептора андрогену) або PR (рецептора прогестерону). Коротко кажучи, випробування конкурентного зв'язування рецепторів стероїдів виконуються у буфері, який містить 20 мМ буфера HEPES (pH=7,6), 0,2 мМ EDTA, 75 мМ NaCl, 1,5 мМ MgCl2, 20 % гліцерину, 20 мМ молібдату натрію, 0,2 мМ DTT (дитіотреїтолу), 20 мкг/мл апротиніну та 20 мкг/мл лейпептину (випробувальний буфер). Як правило, при випробуваннях зв'язування 3 рецепторів стероїдів засотосовують радіомічені ліганди, такі як 0,25 нМ [ H]-альдостерону для 3 3 зв'язування MR, 0,3 нМ [ H]-дексаметазону для зв'язування GR, 0,36 нМ [ H]-метилтриєнолону 3 для зв'язування AR та 0,29 нМ [ H]-метилтриєнолону для зв'язування PR, а також 20 мкг лізату 293-MR, 20 мкг лізату 293-GR, 22 мкг лізату 293-AR або 40 мкг лізату 293-PR на лунку. Випробування звичайно виконуються у 96-лунковому форматі. Конкурентні випробовувані 18 UA 100131 C2 5 10 15 20 25 30 35 40 45 50 55 60 сполуки додають у різних концентраціях в межах від приблизно 0,01 нМ до 10 мкМ. Неспецифічне зв'язування визначають у присутності 500 нМ альдостерону для зв'язування MR, 500 нМ дексаметазону для зв'язування GR або 500 нМ метилтриєнолону для зв'язування AR та PR. Реакційні суміші для визначення зв'язування (140 мкл) інкубують протягом ночі при 4C, потім до кожної реакційної суміші додають 70 мкл охолодженого буфера на основі вугільного пилу та декстрану (який містить 0,75 г деревного вугілля та 0,25 г декстрану на 50 мл випробувального буфера). Планшети перемішують на орбітальному струшувачі протягом 8 хв при 4C. Потім планшети центрифугують при швидкості 3000 об/хв при 4C протягом 10 хв. Потім аліквотну частку реакційної суміші для визначення зв'язування об'ємом 120 мкл переносять у другий 96-лунковий планшет, і додають у кожну лунку 175 мкл сцинтиляційної рідини Wallac Optiphase Hisafe 3. Планшети закривають та інтенсивно струшують на орбітальному струшувачі. Після інкубування протягом 2 год. планшети сканують, застосовуючи лічильник Wallac Microbeta. Одержані дані використовують для обчислення оцінного значення IC50 та відсотка 3 інгібування при концентрації 10 мкМ. Значення Kd для [ H]-альдостерону при зв'язуванні MR, 3 3 3 [ H]-дексаметазону при зв'язуванні GR, [ H]-метилтриєнолону при зв'язуванні AR або [ H]метилтриєнолону при зв'язуванні PR визначають шляхом зв'язування при насиченні. Значення IC50 для сполук перетворюють у значення Ki, застосовуючи рівняння Ченга-Прусоффа (ChengPrusoff). При випробуванні за методикою, описаною у загальних рисах вище, сполуки за Прикладами 1-50 мають Ki10 нМ у випробуваннях зв'язування MR. За варіантом, якому віддається перевага, сполуки за цим винаходом мають Ki5 нМ у випробуваннях зв'язування MR, а варіантом, якому віддається більша перевага, Ki1 нМ. Конкретно, сполуки за Прикладом 1, Прикладом 8 та Прикладом 23 мають Ki у випробуваннях зв'язування MR приблизно 0,79 нМ, 0,08 нМ та 0,23 нМ (вказані середні геометричні значення для n=2), відповідно, демонструючи, таким чином, що сполуки, які охоплюються цим винаходом, є ефективними лігандами людського MR. Функціональні випробування модулювання рецепторів ядерних стероїдних гормонів Фізіологічні ефекти альдостерону виявляються через взаємодію з рецепторами мінералокортикоїдів. Після цитоплазмового зв'язування альдостерону з MR комплекс лігандрецептор переміщується в ядро клітини, де він зв'язується з елементами реакції на гормони у ДНК та ініціює експресію генів-мішеней. Для демонстрування здатності сполук за цим винаходом модулювати активність рецепторів стероїдних гормонів (тобто агонізувати, частково агонізувати, частково антагонізувати або антагонізувати) виконують біопроби, які детектують функціональну модуляцію експресії гена-мішені у клітинах, перехідно трансфікованих протеїном ядерного рецептора та конструктом елемента реакції на гормон з геном-репортером. Розчинники, реагенти та ліганди, застосовувані у функціональних випробуваннях, є легко доступними з комерційних джерел, або вони можуть бути виготовлені фахівцем у цій галузі. A. Скринінг набору рецепторів ядерних гормонів Людські ембріональні ниркові клітини HEK293 трансфікують рецепторами стероїдних гормонів та плазмідами генів-репортерів із використанням прийнятного реагента для трансфікування, такого як Fugene. Коротко кажучи, плазміду репортера, яка містить дві копії промоторів пробазину ARE та TK (тимідинкінази) вище кДНК репортера люциферази, трансфікують у клітини HEK293 з плазмідою, яка конститутивно експресує рецептор людського андрогену (AR), застосовуючи промотор вірусу CMV (цитомегаловірусу). Плазміду репортера, яка містить дві копії промоторів GRE та TK вище кДНК репортера люциферази, трансфікують із плазмідою, яка конститутивно експресує людський рецептор глюкокортикоїду (GR), людський рецептор мінералокортикоїду (MR) або людський рецептор прогестерону (PR), застосовуючи промотор вірусу CMV. Клітини трансфікують у флаконах T150 см у середовищі DMEM з домішкою 5 % очищеної активованим вугіллям сироватки плода корови (FBS). Після інкубування протягом ночі трансфіковані клітини обробляють трипсином, висівають у 96-лункові планшети у середовищі DMEM з домішкою 5 % очищеної активованим вугіллям сироватки плода корови FBS, інкубують протягом 4 год., після чого обробляють різними концентраціями випробовуваних сполук в межах від приблизно 0,01 нМ до 10 мкМ. При випробуванні в режимі антагонізму до середовища додають низькі концентрації антагоніста кожного відповідного рецептора (0,08 нМ альдостерону для MR, 0,25 нМ дексаметазону для GR, 0,66 нМ метилтриєнолону для AR та 0,08 нМ промегестону для PR). Після інкубування протягом 24 год. із випробовуваними сполуками клітини лізують, і визначають активність люциферази, застосовуючи стандартні способи. Одержані результати апроксимують чотирипараметричною логістичною кривою для 19 UA 100131 C2 5 10 15 20 25 30 35 40 45 50 55 60 визначення EC50. Відсоткову ефективність (для сполук з насиченими максимальними реакціями) або відсоток максимальної стимуляції (для сполук з максимальними реакціями, які не насичуються) визначають відносно максимальної стимуляції, одержаної при застосуванні таких агоністів порівняння: 30 нМ альдостерону для MR, 100 нМ метилтриєнолону для AR, 30 нМ промегестону для PR та 100 нМ дексаметазону для GR. Аналогічним чином визначають IC50, використовуючи дані випробувань в режимі антагонізму. При випробуваннях в режимі антагонізму відсоток інгібування визначають шляхом зіставлення активності випробовуваної сполуки у присутності низької концентрації агоніста (0,08 нМ альдостерону для MR, 0,25 нМ дексаметазону для GR, 0,66 нМ метилтриєнолону для AR та 0,08 нМ промегестону для PR) з реакцією, спричиненою тією самою низькою концентрацією агоніста при відсутності випробовуваної сполуки. B. Випробування конкурентних антагоністів hMR Людські ембріональні ниркові клітини HEK293 трансфікують людським MR з використанням тих самих реагентів для трансфікування, плазмід, промоторів, конструктів репортерів, буферів та із застосуванням методик, які описані вище у параграфі "Скринінг набору рецепторів ядерних гормонів" трансфіковані клітини обробляють трипсином, висівають у 96-лункові планшети у середовищі DMEM, яке містить 5 % очищеної активованим вугіллям сироватки плода корови FBS, інкубують протягом 4 год., після чого обробляють різними концентраціями (10 розведень) альдостерону (в межах від приблизно 0,001 нМ до 0,03 мкМ). Здатність альдостерону агонізувати hMR визначають за відсутності та у присутності фіксованих концентрацій випробовуваних сполук; моніторинг здійснюють шляхом вимірювання активності люциферази за допомогою стандартних способів. Потім можна визначити Kb випробовуваних сполук, застосовуючи аналіз за Шильдом (Schild), шляхом побудови логарифмічної кривої (відношення доз - 1) як функції логарифма концентрації антагоніста з використанням рівняння: Log (DR 1)=Log [антагоніст] - Log Kb, де відношення доз (DR) означає відношення EC50 альдостерону у присутності випробовуваної сполуки до EC50 альдостерону за відсутності випробовуваної сполуки. При випробуванні за методикою, описаною у загальних рисах вище, сполуки за цим винаходом, взяті за приклади, мають Kb200 нМ у випробуваннях конкурентних антагоністів MR. За варіантом, якому віддається перевага, сполуки за цим винаходом мають K b50 нМ у випробуваннях конкурентних антагоністів MR, а варіантом, якому віддається більша перевага, Kb10 нМ. Конкретно, сполуки за Прикладом 1, Прикладом 8 та Прикладом 23 мають у випробуваннях конкурентних антагоністів MR значення Kb приблизно 1,2 нМ, 1,1 нМ та 2,5 нМ (вказані середні геометричні значення для n=2), відповідно, демонструючи, таким чином, що сполуки, які охоплюються цим винаходом, є ефективними антагоністами людського MR. Модель опосередкованого альдостероном захворювання нирок іn vivo Самців пацюків лінії Sprague Dawley (маса тіла 240-280 г), підданих односторонній нефректомії, поміщали окремо протягом одного тижня при вільному доступі до водопровідної води та корму для гризунів 5001. Після акліматизації збирали проби сечі протягом 24 год., та аналізували на загальний вміст протеїнів та креатиніну у сечі для визначення базового рівня. Тварин розподіляли по дослідних групах за випадковим принципом на основі маси тіла та базового рівня протеїну в сечі. Відбирали проби базової сироватки крові шляхом надрізання хвоста, та аналізували згадані проби на вміст сечовинного азоту в крові (BUN), креатиніну та електролітів. Після відбирання базових проб усіх пацюків, за винятком контрольної групи, протягом дослідження утримували на кормі, який містив 6 % солі, та питній воді із вмістом 0,3 % KCl. Контрольних тварин протягом дослідження утримували на кормі 5001 та водопровідній воді, і не вводили їм альдостерон. Тваринам неконтрольних груп (тобто груп, які одержували досліджувану сполуку, та груп, які одержували тільки носій) імплантували під ізофлурановою анестезією міні-насоси Alza для підшкірного введення 2,5 мкл/год. d-альдостерону в 0,01 % DMSO при швидкості 0,75 мкг/год. протягом 28 діб. Починаючи з наступної доби після імплантування міні-насосів для введення альдостерону, тваринам вводили шляхом щоденного одноразового примусового згодовування (10 мл/кг) випробовувану сполуку у носії, який містив 1 % карбоксиметилцелюлози (CMC)/0,25 % полісорбату 80, або тільки носій. Через 2 тижні та 4 тижні після введення сполуки або тільки носія повторно збирали проби сечі, та аналізували їх на загальний вміст протеїнів та креатиніну. Наприкінці дослідження відбирали фармакокінетичні проби для 8 моментів часу (0,5 год., 1 год., 2 год., 3 год., 6 год., 8 год., 12 год. та 24 год.). Крім того, серця та нирки видаляли та фіксували у формаліні з 10 % буфера для забарвлення гематоксиліном та еозином (H&E) і трихромом Массона (Masson) для виявлення структурних порушень у серцевих та ниркових тканинах. Наприкінці дослідження відбирали сироватку крові шляхом серцевої пункції для додаткового аналізу на сироватковий BUN, креатинін та 20 UA 100131 C2 5 10 15 20 25 30 35 40 45 50 55 60 електроліти. За методикою, описаною у загальних рисах вище, сполуки за Прикладом 1, Прикладом 8 та Прикладом 23 при введенні в кількості 10 мг/кг на добу протягом 14 діб забезпечували зниження виділення протеїнів із сечею на приблизно 49 мг/доба, 83 мг/доба та 64 мг/доба (середні значення для n=8 для кожної сполуки), відповідно, у порівнянні з показниками для тварин, які одержували носій, демонструючи, таким чином, що сполуки, які охоплюються цим винаходом, мають високу нефропротекторну активність in vivo. Для підтвердження того, що сполука має знижену здатність до спричинення гіперкаліємії або знижену ймовірність її виникнення, можна використати описану нижче модель. Випробування на модулювання електролітів in vivo Самців пацюків лінії Sprague Dawley (маса тіла 240-280 г) піддавали адреналектомії, після чого утримували на кормі для гризунів 5001 та питному 1 % розчині NaCl протягом 6 діб після операції. Потім тварин витримували без годування протягом ночі, а 1 % сольовий питний розчин замінювали водопровідною водою з вільним доступом до неї. У день дослідження голодних тварин зранку розподіляли для лікування за випадковим принципом на основі маси тіла після голодування. Контрольним тваринам (тобто тим, які не одержували альдостерону або випробовуваної сполуки) вводили 10 мл/кг носія випробовуваної сполуки, який містив 0,5 % CMC/0,25 % полісорбату 80/2,7 % NaCl, шляхом примусового згодовування та 1 мл/кг носія альдостерону (0,01 % DMSO у воді) шляхом підшкірного впорскування. Тваринам групи, яка одержувала носій, вводили той самий носій випробовуваної сполуки шляхом примусового згодовування та альдостерон 3 мкг/кг підшкірно. Випробовувані сполуки суспендували у носії, який містив карбоксиметилцелюлозу та NaCl. Тварини, яких обробляли випробовуваною сполукою, одержували випробовувану сполуку, суспендовану в носії, який містив карбоксиметилцелюлозу та NaCl, та альдостерон 3 мкг/кг підшкірно. Одразу після введення тварин вміщували у бокси для дослідження метаболізму з вільним доступом до водопровідної води. Після введення доз збирали проби сечі протягом 5 год., і досліджували виведення електролітів. Результати представлені у формі логарифма відношення виведення Na/K. Сполуки можна випробовувати у різних дозах, і таким чином визначати, чи стимулює певна сполука підвищення відношення Na/K у сечі (показника підвищеної концентрації калію у сироватці крові). Вважається, що фахівець у цій галузі може без здійснення додаткових винахідницьких кроків використати поданий вище опис для здійснення цього винаходу у найповнішому обсязі. Подані нижче розділи "Підготовчі синтези" та "Приклади" наведені для більш детального ілюстрування винаходу та представлення типових способів синтезу сполук формули (I). Однак ці описи не призначені для обмеження жодним чином обсягу винаходу. Реагенти та вихідні матеріали є легко доступними для фахівця у цій галузі або можуть бути легко синтезовані ним. Для фахівців у цій галузі будуть зрозумілі відповідні відхилення від методик, описаних у прикладах. Назви сполук за цим винаходом, як правило, утворені за допомогою програми ChemDraw Ultra , версія 10.0. Підготовчий синтез 1 1-бром-4-фтор-2-(2-йодбензилокси)бензол Перемішують суміш 2-йодбензилброміду (0,29 моль, 90 г), 2-бром-5-фторфенолу (0,29 моль, 57,9 г) та карбонату калію (0,46 моль, 63 г) у N, N-диметилформаміді (750 мл) при кімнатній температурі протягом 16 год. Додають воду (1 л), перемішують одержану суміш протягом 1 год., відділяють тверду речовину фільтруванням, промивають водою, і сушать у вакуумній сушильній 1 шафі (20 мм рт.ст./60C), одержуючи вказану в заголовку сполуку (121 г, >100 %). H ЯМР (400 МГц, DMSO-d6) 5,11 (s, 2H), 6,81 (t, 1H), 7,13 (t, 1H), 7,19 (dd, 1H), 7,46 (t, 1H), 7,59 (d, 1H), 7,62 (t, 1H), 7,93 (d, 1H). Підготовчий синтез 2 Етиловий складний ефір 3-[2-(2-бром-5-фторфеноксиметил)феніл]акрилової кислоти До суміші 1-бром-4-фтор-2-(2-йодбензилокси)бензолу (0,29 моль, 117,4 г), ацетату натрію (0,44 моль, 36,1 г, 1,5 екв.), броміду тетра-н-бутиламонію (0,29 моль, 90,3 г), ацетату паладію (II) (8 ммоль, 1,8 г, 3 моль-%) та N-метилпіролідинону (900 мл) при 55-60C додають краплями розчин етилакрилату (0,32 моль, 34,3 мл) у N-метилпіролідиноні (200 мл). Охолоджують реакційну суміш до кімнатної температури, і додають воду (2 л) та метил-трет-бутиловий складний ефір (2 л). Фільтрують через діатомову землю, додають етилацетат (1 л), розділяють шари, і промивають водою (2 л). Органічну фазу сушать над безводним сульфатом натрію, фільтрують, та концентрують. Одержану тверду речовину суспендують у гексані (1 л), витримують у холодильнику протягом 2 год., фільтрують, і промивають холодним гексаном (500 мл). Сушать у вакуумній сушильній шафі (50C та 20 мм рт.ст.), і одержують вказану в 21 UA 100131 C2 5 10 15 20 25 30 заголовку сполуку у вигляді блідо-жовтої твердої речовини (104,4 г, 95 %). LC-MS m/z 381,0 + [M+H] . Підготовчий синтез 3 Етиловий складний ефір (E)-(3-фтор-6H-дибензо[b, e]оксепін-11-іліден)оцтової кислоти Суміш етилового складного ефіру 3-[2-(2-бром-5-фторфеноксиметил)феніл]акрилової кислоти (0,25 моль, 94 г), ацетату натрію (0,37 моль, 30 г, 1,5 екв.), броміду тетра-нбутиламонію (0,25 моль, 81 г) та ацетату паладію (II) (7 ммоль, 1,7 г, 3 моль-%) в Nметилпіролідиноні (850 мл) нагрівають до 100-110C та витримують при цій температурі протягом 6 год. Охолоджують до кімнатної температури, розводять водою (1 л), фільтрують через діатомову землю, і промивають етилацетатом (2 л). Переносять фільтрат у ділильну лійку, додають воду (500 мл), і розділяють шари. Органічний шар промивають водою (21,5 л), сушать над безводним сульфатом натрію, фільтрують через шар діоксиду кремнію, промивають етилацетатом (1,5 л), і концентрують досуха. До одержаної твердої речовини додають гексан (1 л), витримують у холодильнику протягом 2 год., фільтрують, промивають гексаном (500 мл), і сушать при 50C та 20 мм рт.ст., одержуючи вказану в заголовку сполуку (64,3 г, 87 %). LC-MS + m/z 299,0 [M+H] . Підготовчий синтез 4 (E)-11-бромметилен-3-фтор-6,11-дигідродибензо[b, e]оксепін До суспензії етилового складного ефіру (3-фтор-6H-дибензо[b, e]оксепін-11-іліден)оцтової кислоти (0,23 моль, 69,5 г) в ізопропанолі (725 мл) додають розчин гідроксиду літію (0,53 моль, 12,0 г) у воді (125 мл), нагрівають одержану суміш до 70C та витримують при цій температурі протягом 4 год. Дають суміші охолодитися до 40C, після чого обробляють льодяною оцтовою кислотою (0,44 моль, 25 мл). Після перемішування протягом 15 хв додають N-бромсукцинимід (0,25 моль, 44 г). Суміш спінюється, температура підвищується до 45C, і через кілька хвилин утворюється тверда речовина. Суміш перемішують при 40-45C протягом години, і охолоджують до кімнатної температури. Додають розчин бісульфіту натрію (4,5 г) у воді (150 мл), насичений водний розчин бікарбонату натрію (150 мл) та воду (450 мл). Одержану суспензію фільтрують, і промивають охолодженою сумішшю ізопропанолу з водою (1:1, 300 мл). Тверду речовину сушать при 60C та 20 мм рт.ст. протягом ночі, і одержують вказану в заголовку сполуку (65,8 г, 1 93 %). H ЯМР (400 МГц, DMSO-d6) 4,9-5,4 (br d, 2H), 6,63 (dd, 1H), 6,77 (dt, 1H), 7,13 (s, 1H), 7,32-7,46 (m, 4H), 7,52 (dd, 1H). Підготовчий синтез 5 (E)-2-((3-фтордибензо[b, e]оксепін-11(6H)-іліден)метил)-4,4,5,5-тетраметил-1,3,2діоксаборолан O F O B O 35 40 45 50 До перемішуваної суміші (E)-11-(бромметилен)-3-фтор-6,11-дигідродибензо[b, e]-оксепіну (49 ммоль, 15 г) та біс(пінаколато)дибору (64 ммоль, 16 г) в 1,4-діоксані (250 мл) додають ацетат калію (150 ммоль, 15 г). Продувають суміш азотом, додають адукт дихлор[1,1'біс(дифенілфосфіно)фероцен]паладію (II) із дихлорметаном (2,46 ммоль, 1,80 г), нагрівають до 65C та витримують при цій температурі протягом ночі. Охолоджують до кімнатної температури, фільтрують через діатомову землю, промивають етилацетатом, і концентрують фільтрат у вакуумі. Додають метанол (200 мл), і перемішують суміш протягом 1 год. у роторному випарнику без вакуумування, спричинюючи утворення коричневої твердої речовини. Одержані темнокоричневі кристали відділяють фільтруванням, і сушать у вакуумі протягом ночі, одержуючи вказану в заголовку сполуку (7,28 г, 42 %). Фільтрат концентрують, і очищають залишок хроматографією на колонці з елююванням градієнтом від 0 % до 16 % етилацетату в гексані, одержуючи вказану в заголовку сполуку у вигляді жовтої твердої речовини (3,46 г, 1 20 %). Загальний вихід по реакції 10,7 г (62 %). H ЯМР (300 МГц, CDCl3) 7,36 (dd, J=8,8 Гц, 6,8 Гц, 1H), 7,32-7,27 (m, 4H), 6,64-6,57 (m, 1H), 6,48 (dd, J=10,3 Гц, 2,6 Гц, 1H), 5,98 (s, 1H), 5,20 (br s, 1H), 1,15 (s, 12H). Підготовчий синтез 6 22 UA 100131 C2 5 10 15 20 25 30 35 40 45 50 55 1-бром-3-фторбензальдегід До розчину діізопропіламіну (2,27 моль, 320 мл) у безводному тетрагідрофурані (800 мл) у колбі місткістю 5 л додають краплями при 0C 1,6 M розчин бутиллітію (1,16 л, 1,86 моль) у гексані протягом 1,5 год. Перемішують одержаний жовтий розчин при 0C протягом 30 хв. В окремій колбі місткістю 12 л розчиняють 1-бром-3-фторбензол (203 мл, 1,86 моль) у безводному тетрагідрофурані (650 мл), і охолоджують до -78C. Заздалегідь виготовлений розчин LDA переносять за допомогою канюлі у завантажувальну лійку, і додають краплями до розчину 1бром-3-фторбензолу протягом 2 год. так, щоб температура не піднімалася вище -70C. Одержану суспензію перемішують при -78C протягом 30 хв. Додають краплями розчин Nметил-N-фенілформаміду (230 мл, 1,86 моль, 1,00 екв.) у безводному тетрагідрофурані (1,15 л) при -78C протягом 1 год., підтримуючи температуру ніжче -70C. Перемішують холодну реакційну суміш протягом 2 год., потім дозволяють їй нагріватися до кімнатної температури протягом ночі. Розводять реакційну суміш метил-трет-бутиловим складним ефіром (3 л), гасять 1 M хлористоводневою кислотою (4 л), та інтенсивно перемішують протягом 3 год. Розділяють шари, та екстрагують водний шар метил-трет-бутиловим складним ефіром (1 л). Промивають об'єднані органічні фази 1 M хлористоводневою кислотою (21 л), водою (1 л), розсолом (1 л), сушать над сульфатом магнію, фільтрують, та концентрують, одержуючи оранжеве масло, яке 1 повільно твердне, утворюючи вказану в заголовку сполуку (394 г, 105 %). H ЯМР (CDCl3) 10,37 (s, 1H), 7,50 (d, 1H), 7,44 (m, 1H), 7,17 (t, 1H). Підготовчий синтез 7 1-бром-3-фторбензиловий спирт До розчину 1-бром-3-фторбензальдегіду (1,60 кг, 7,91 моль) у метанолі (14 л) при 0C додають частинами боргідрид натрію (284 г, 7,93 моль) протягом 30 хв, регулюючи виділення тепла та газу. Через 15 хв гасять водою (500 мл), і концентрують, одержуючи жовте масло. Розчиняють це масло в етилацетаті (6 л), і промивають водою (3 л). Екстрагують водний шар етилацетатом (1 л). Промивають об'єднані органічні фази розсолом (2 л), сушать над сульфатом натрію, фільтрують, та концентрують, одержуючи вказану в заголовку сполуку у 1 вигляді оранжевого масла (1614 г, 100 %). H ЯМР (CDCl3) 7,40 (d, 1H), 7,18 (m, 1H), 7,07 (t, 1H), 4,86 (s, 2H). Підготовчий синтез 8 1-бром-3-фторбензилбромід До розчину 1-бром-3-фторбензилового спирту (1,61 кг, 7,85 моль) у хлороформі (14 л) при 0C додають однією порцією піридин (770 мл, 9,52 моль) (вивільнюється незначна кількість тепла), і перемішують при 0C протягом 5 хв. Додають краплями трибромід фосфору (900 мл, 9,49 моль), підтримуючи температуру в масі нижче 20C, і перемішують одержаний розчин протягом ночі, даючи йому нагріватися до кімнатної температури. Охолоджують реакційну суміш до 0C, і повільно гасять льодяною водою (2 л). Переносять суміш у скляний реактор місткістю 50 л, розділяють шари, та екстрагують водний шар хлороформом (1 л). Промивають об'єднані органічні фази 5 % сірчаною кислотою (2 л), насиченим водним розчином бікарбонату натрію (2 л), розсолом (2 л), сушать над сульфатом магнію, фільтрують, та концентрують, 1 одержуючи вказану в заголовку сполуку увигляді жовтого масла (1753 г, 83 %). H ЯМР (CDCl3) 7,43 (d, 1H), 7,21 (m, 1H), 7,09 (t, 1H), 4,68 (s, 2H). Підготовчий синтез 9 1-бром-2-(2-бром-6-фтор-бензилокси)-4-фторбензол До розчину 1-бром-3-фторбензилброміду (1,75 кг, 6,52 моль) та 2-бром-5-фторфенолу (730 мл, 16,56 моль, 1,01 екв.) у N, N-диметилформаміді (14 л) при 0C додають однією порцією карбонат калію (1,36 кг, 9,80 моль), і перемішують одержану суспензію при охолодженні протягом 1 год., а потім при кімнатній температурі протягом ночі. Переносять реакційну суміш у скляний реактор місткістю 50 л, додають краплями воду (14 л) протягом 30 хв та періодично лід для регулювання вивільнення тепла кристалізації. Перемішують при кімнатній температурі протягом 1 год., відділяють злегка забарвлену тверду речовину фільтруванням, промивають водою, і сушать протягом ночі при 50C, одержуючи вказану в заголовку сполуку (2,22 кг, 90 %). 1 H ЯМР (CDCl3) 7,51 (m, 2H), 7,30 (m, 1H), 7,14 (t, 1H), 6,89 (dd, 1H), 6,67 (m, 1H), 5,26 (d, 2H). Підготовчий синтез 10 Етиловий складний ефір (E)-(3,7-дифтор-6H-дибензо[b, e]оксепін-11-іліден)оцтової кислоти Знегажують суміш 1-бром-2-(2-бром-6-фтор-бензилокси)-4-фторбензолу (886 ммоль, 335 г), етилакрилату (931 ммоль, 101 мл, 1,05 екв.), Pd(OAc)2 (22,2 ммоль, 4,97 г), броміду тетрабутиламонію (886 ммоль, 286 г) та ацетату натрію (4,40 моль, 363,5 г) у Nметилпіролідиноні (3,3 л) шляхом п'ятиразового почергового вакуумування та заповнення 23 UA 100131 C2 5 10 15 20 25 30 35 40 45 50 55 азотом. Нагрівають одержану суміш до 120C протягом 2 год. Охолоджують до 35C, та додають воду (5,5 л) протягом 40 хв. Відділяють осад фільтруванням, промивають водою (2500 мл), і сушать на повітрі протягом 30 хв. Повторно суспендують в ізопропанолі (1 л) протягом 2 год., та охолоджують на льоду протягом 1 год. Відділяють темносіру тверду речовину фільтруванням, промивають холодним ізопропанолом (5100 мл), і сушать при 50C у вакуумі, одержуючи вказану в заголовку сполуку (191 г, 68 %). Підготовчий синтез 11 (E)-11-бромметилен-3,7-дифтор-6,11-дигідро-дибензо[b, e]оксепін Нагрівають при перемішуванні суспензію етилового складного ефіру (E)-(3,7-дифтор-6H-дибензо[b, e]оксепін-11-іліден)оцтової кислоти (601 ммоль, 190 г) у метанолі (1,9 л) та 5 н гідроксид натрію (1,20 моль, 240 мл) до 50C протягом 2 год. Видаляють метанол у вакуумі, та повторно суспендують одержану чорну масу у воді (3 л). Додають 5 н хлористоводневу кислоту (250 мл) протягом 45 хв, перемішують протягом 2,5 год., фільтрують, і промивають залишок водою (4200 мл). Сушать сіру тверду речовину у вакуумі при 60C протягом ночі та при 80C протягом 6 год. Розчиняють твердий продукт (ще вологий, містить приблизно 95 г води) та ацетат літію (60,1 ммоль, 4 г, 0,1 екв.) в ацетонітрилі (1,7 л), і перемішують протягом 15 хв. Додають однією порцією N-бромсукцинимід (661 ммоль, 118 г), і перемішують протягом 2,5 год. Додають 0,25 M розчин тіосульфату натрію (400 мл), потім насичений водний розчин бікарбонату натрію (400 мл) та воду (900 мл). Перемішують суміш протягом 1 год. при кімнатній температурі, а потім перемішують протягом 2 год. на льодяній бані. Фільтрують суміш, і промивають холодним ацетонітрилом (300 мл). Сушать протягом ночі у вакуумі при 50C, і одержують вказану в заголовку сполуку у вигляді світло-бежевої твердої 1 речовини (173,3 г, 89 %). H ЯМР (DMSO-d6) 5,25 (s, 2H), 6,67 (dd, 1H), 6,81 (dt, 1H), 7,20 (s, 1H), 7,22 (d, 1H), 7,28 (t, 1H), 7,36 (dd, 1H), 7,48 (m, 1H). Підготовчий синтез 12 (E)-2-((3,7-дифтордибензо[b, e]оксепін-11(6H)-іліден)метил)-4,4,5,5-тетраметил-1,3,2діоксаборолан До суміші (E)-11-(бромметилен)-3,7-дифтор-6,11-дигідродибензо[b, e]оксепіну (46,4 ммоль, 15,0 г) та біс(пінаколато)дибору (50,3 ммоль, 15,3 г) у 1,4-діоксані (150 мл) додають ацетат натрію (74,3 ммоль, 7,29 г), трициклогексилфосфін (6,03 ммоль, 1,69 г) та Pd2(dba)3 (2,32 ммоль, 2,13 г). Продувають суміш азотом шляхом барботування, потім перемішують при 65C протягом ночі. Після охолодження до кімнатної температури фільтрують суміш через шар діатомової землі, промивають цей шар етилацетатом, та видаляють розчинник, одержуючи коричневе масло. Це масло розводять метанолом (180 мл), і перемішують одержану суміш у роторному випарнику без вакуумування протягом 3 год., внаслідок чого утворюється білий осад. Цей осад відділяють вакуум-фільтруванням, і одержують вказану в заголовку сполуку (10,8 г, 63 %) у 1 вигляді білої твердої речовини. H ЯМР (300 МГц, CDCl3) 7,34 (dd, J=8,8 Гц, 6,7 Гц, 1H), 7,247,18 (m, 1H), 7,08-7,01 (m, 2H), 6,65-6,59 (m, 1H), 6,50 (dd, J=10,3 Гц, 2,6 Гц, 1H), 5,99 (s, 1H), 5,31 (d, J=0,9 Гц, 2H), 1,16 (s, 12H). Підготовчий синтез 13 (E)-11-бромметилен-3,8-дифтор-6,11-дигідро-дибензо[b, e]оксепін Вказану в заголовку сполуку одержують, як описано у Підготовчому синтезі 9, Підготовчому синтезі 10 та Підготовчому синтезі 11, використовуючи як вихідні сполуки 1-бром-2-бромметил4-фторбензол та 2-бром-5-фторфенол. Підготовчий синтез 14 (E)-2-((3,8-дифтордибензо[b, e]оксепін-11(6H)-іліден)метил)-4,4,5,5-тетраметил-1,3,2діоксаборолан До суміші (E)-11-(бромметилен)-3,8-дифтор-6,11-дигідродибензо[b, e]оксепіну (90,0 ммоль, 29,0 г) та біс(пінаколато)дибору (117 ммоль, 29,5 г) у 1,4-діоксані (295 мл) додають ацетат натрію (144 ммоль, 14,1 г), трициклогексилфосфін (11,7 ммоль, 3,28 г) та Pd2(dba)3 (4,14 ммоль, 3,80 г) Продувають суміш азотом шляхом барботування, потім перемішують при 65C протягом ночі. Після охолодження до кімнатної температури фільтрують суміш через шар діатомової землі, і промивають цей шар етилацетатом. Концентрують фільтрат у вакуумі, одержуючи коричневе масло. Це масло розводять метанолом (180 мл), і перемішують одержану суміш у роторному випарнику без вакуумування протягом 1,5 год., внаслідок чого утворюється коричневий осад. Цей осад відділяють вакуум-фільтруванням, і одержують вказану в заголовку 1 сполуку у вигляді коричневої твердої речовини (24,2 г, 73 %). H ЯМР (300 МГц, CDCl3) 7,35 (dd, J=8,8 Гц, 6,7 Гц, 1H), 7,29-7,24 (m, 1H), 7,05-6,93 (m, 2H), 6,65-6,59 (m, 1H), 6,49 (dd, J=10,2 Гц, 2,6 Гц, 1H), 5,99 (s, 1H), 5,15 (br s, 2H), 1,16 (s, 12H). 60 24 UA 100131 C2 5 10 15 20 25 30 35 40 45 Підготовчий синтез 15 2-трет-бутоксикарбоніламінопропіловий складний ефір (R)-метансульфонової кислоти Додають краплями метансульфонілхлорид (0,128 моль, 14,7 г, 1,5 екв.) до розчину (R)-(+)-2трет-бутоксикарбоніламіно-1-пропанолу (0,085 моль, 15,0 г) та триетиламіну (0,12 моль, 17,3 г, 2,0 екв.) у дихлорметані (150 мл) при 0C в атмосфері азоту. Після завершення додавання підігрівають реакційну суміш до кімнатної температури, та перемішують протягом 2 год. Розводять реакційну суміш дихлорметаном, та промивають 0,1 н хлористоводневою кислотою та насиченим розчином бікарбонату натрію. Сушать органічний шар над безводним сульфатом натрію, фільтрують, та концентрують у вакуумі, одержуючи вказану в заголовку сполуку (20,8 г, 1 96 %) у вигляді білої твердої речовини. H ЯМР (400 МГц, CDCl3) 1,24 (d, 3H), 1,43 (s, 9H), 3,03 (s, 3H), 3,97 (br s, 1H), 4,14 (dd, 1H), 4,21 (br s, 1H), 4,64 (br s, 1H, Boc-NH). Підготовчий синтез 16 Трет-бутиловий складний ефір (R)-(1-метил-2-морфолін-4-іл-етил)карбамінової кислоти Нагрівають суміш 2-трет-бутоксикарбоніламінопропілового складного ефіру (R)метансульфонової кислоти (0,083 моль, 20 г) та морфоліну (0,83 моль, 72,2 г) в ацетонітрилі (210 мл) до 65C протягом 16 год. Концентрують реакційну суміш, розводять залишок водою, екстрагують дихлорметаном, органічні шари об'єднують, та промивають їх водою. Сушать органічний шар над безводним сульфатом натрію, фільтрують, та концентрують у вакуумі, 1 одержуючи вказану в заголовку сполуку (12 г, 62 %) у вигляді густого жовтого масла. H ЯМР (400 МГц, CDCl3) 1,13 (d, 3H), 1,43 (s, 9H), 2,21 (dd, 1H), 2,26-2,32 (m, 1H), 2,35-2,39 (m, 2H), 2,48-2,55 (m, 2H), 3,63-3,70 (m, 5H), 4,68 (br s, 1H, -NH). Підготовчий синтез 17 Гідрохлорид (R)-1-метил-2-морфолін-4-іл-етиламін Додають 1,6 M розчин хлороводню в діоксані (120 мл) до розчину трет-бутилового складного ефіру (R)-(1-метил-2-морфолін-4-іл-етил)карбамінової кислоти (0,05 моль, 12 г) у безводному дихлорметані (75 мл) при 0C. Нагрівають реакційну суміш до кімнатної температури, та перемішують її протягом 2 год. Осад відділяють фільтруванням, і сушать у вакуумі, одержуючи 1 вказану в заголовку сполуку (9,2 г, 86 %) у вигляді білої твердої речовини. H ЯМР (400 МГц, DMSO-d6) 1,29 (d, 3H), 3,10 (br s, 2H), 3,38 (br s, 4H), 3,79 (br s, 5H). Підготовчий синтез 18 (R)-(4-бром-2-нітрофеніл)-(1-метил-2-морфолін-4-іл-етил)амін Суміш гідрохлориду (R)-1-метил-2-морфолін-4-іл-етиламіну (0,04 моль, 8,7 г), 5-бром-2фторнітробензолу (0,048 моль, 10,5 г), триетиламіну (0,20 моль, 20,2 г) та диметиламінопіридину (0,0004 моль, 0,048 г) в етилацетаті (220 мл) нагрівають зі зворотним холодильником протягом 16 год. Охолоджують реакційну суміш до кімнатної температури, розводять етилацетатом, та промивають водою та розсолом. Сушать органічний шар над безводним сульфатом натрію, фільтрують, та концентрують. Очищають залишок на колонці із силікагелем, використовуючи як елюент суміш 10 % етилацетату з гексаном, і одержують 1 вказану в заголовку сполуку (12,8 г, 95 %) у вигляді оранжевої твердої речовини. H ЯМР (400 МГц, CDCl3) 1,29 (d, 3H), 2,47-2,51 (m, 5H), 2,53-2,58 (m, 1H), 3,64-3,69 (m, 4H), 3,72-3,78 (m, 1H), 6,78 (d, 1H), 7,46 (dd, 1H), 8,30 (d, 1H), 8,33 (d, 1H). Проміжні продукти, перелічені у поданій нижче таблиці, одержують загалом за методиками, описаними для Підготовчого синтезу 15, Підготовчого синтезу 16, Підготовчого синтезу 17 та Підготовчого синтезу 18, використовуючи як вихідні сполуки відповідно (S)-(-)-2-третбутоксикарбоніламіно-1-пропанол або трет-бутил-N-(2-гідроксіетил)карбамат. Підг. синт. 19 50 55 Хімічна назва Фізичні дані (S)-(4-бром-2-нітро-феніл)- 1H ЯМР (400 МГц, CDCl3) 1,29 (d, 3H), 2,46-2,58 (m, (1-метил-2-морфолін-46H), 3,63-3,69 (m, 4H), 3,71-3,79 (m, 1H), 6,77 (d, 1H), ілетил)амін 7,45 (dd, 1H), 8,29 (d, 1H), 8,33 (d, 1H) Підготовчий синтез 20 (4-бром-2-нітрофеніл)-(4-метилпіперазин-1-іл)амін Витримують суміш 1-аміно-4-метилпіперазину (0,217 моль, 25,0 г), 5-бром-2фторнітробензолу (0,239 моль, 52,6 г) та триетиламін (0,46 моль, 46 г) в етилацетаті (900 мл) при нагріванні зі зворотним холодильником протягом 16 год. Охолоджують реакційну суміш до кімнатної температури, розводять етилацетатом, та промивають водою та розсолом. Сушать органічний шар над безводним сульфатом натрію, фільтрують, та концентрують. Очищають залишок на колонці із силікагелем, використовуючи як елюент суміш 5 % метанолу з + дихлорметаном, і одержують вказану в заголовку сполуку (43,2 г., 63 %). ES-MS m/z 315 [M+1] . 25 UA 100131 C2 5 10 15 20 25 30 35 40 45 50 55 60 Підготовчий синтез 21 трет-бутиловий складний ефір 3-(4-бром-2-нітрофеніламіно)азетидин-1-карбонової кислоти Розчиняють 5-бром-2-фторнітробензол (28,8 ммоль, 3,55 мл), трет-бутиловий складний ефір 3-аміноазетидин-1-карбонової кислоти (28,8 ммоль, 4,96 г) та триетиламін (35,9 ммоль, 5,00 мл) в етилацетаті (100 мл), і нагрівають зі зворотним холодильником в атмосфері азоту протягом 48 год. Охолоджують до кімнатної температури. Промивають двічі 0,5 M хлористоводневою кислотою, сушать над безводним сульфатом натрію, фільтрують, та концентрують у вакуумі, 79 81 одержуючи яскраво-оранжеву тверду речовину (11,04 г, >100 %). LC-MS m/z ( Br/ Br) + 394,0/396,0 [M+H] . Підготовчий синтез 22 Гідрохлорид азетидин-3-іл-(4-бром-2-нітрофеніл)аміну Перемішують розчин трет-бутилового складного ефіру 3-(4-бром-2-нітрофеніламіно)азетидин-1-карбонової кислоти (29,66 ммоль, 11,04 г) у 4,0 M розчині хлороводню в діоксані (40 мл) при кімнатній температурі в атмосфері азоту протягом ночі. З розчину випадає тверда речовина. Розводять суміш діетиловим ефіром (200 мл), відділяють тверду речовину фільтруванням, промивають великою кількістю діетилового ефіру, та сушать (8,54 г, 93 %). 1 H ЯМР (400 МГц, CD3OD): 4,12 (dd, 2H), 4,45 (dd, 2H), 4,72 (m, 1H), 6,76 (d, 1H), 7,62 (dd, 1H), 79 81 + 8,30 (d, 1H). LC-MS m/z ( Br/ Br) 272,0/274,0 [M+H] . Підготовчий синтез 23 (4-бром-2-нітрофеніл)-(1-метилазетидин-3-іл)амін До суміші гідрохлориду азетидин-3-іл-(4-бром-2-нітрофеніл)аміну (27,7 ммоль, 8,54 г) в ацетонітрилі (100 мл) та 37 % водного розчину формальдегіду (83,0 ммоль, 6,28 мл, 3,00 екв.) при 0C повільно додають триацетоксиборгідрид натрію (88,6 ммоль, 18,8 г). Перемішують при кімнатній температурі в атмосфері азоту протягом ночі. Видаляють леткі компоненти під зниженим тиском. Розводять етилацетатом, промивають двічі 10 % водним розчином бікарбонату натрію, сушать над безводним сульфатом натрію, фільтрують, та концентрують під зниженим тиском, одержуючи 8,5 г оранжевої твердої речовини. Очищають цю речовину на колонці зі 120 г діоксиду кремнію при елююванні сумішшю 5 % метанолу з дихлорметаном, і 79 81 + одержують вказану в заголовку сполуку (5,92 г, 75 %). LC-MS m/z ( Br/ Br) 286,0/288,0 [M+H] . Підготовчий синтез 24 2-метиловий складний ефір 1-трет-бутилового складного ефіру (2S, 4R)-4-гідроксипіролідин1,2-дикарбонової кислоти Додають краплями тіонілхлорид (1,07 моль, 128,2 г) до розчину (2S, 4R)-4-гідроксипіролідин2-карбонової кислоти (0,70 моль, 93,0 г) у безводному метанолі (2 л) при 0C в атмосфері азоту. Після закінчення додавання нагрівають реакційну суміш до кімнатної температури, та перемішують протягом 6 год. Концентрують реакційну суміш під зниженим тиском, і одержують гідрохлорид відповідного метилового складного ефіру. До розчину гідрохлориду метилового складного ефіру у безводному дихлорметані (2 л) при 0C додають триетиламін (1,56 моль, 157,5 г), та перемішують протягом 30 хв. Потім додають послідовно N, N-диметиламінопіридин (0,10 моль, 13 г) та ди-трет-бутилдикарбонат (0,85 моль, 185,7 г). підігрівають реакційну суміш до кімнатної температури, та перемішують протягом 18 год. Гасять реакційну суміш водою, відділяють органічний шар, та промивають його водою та насиченим розчином бікарбонату натрію. Сушать органічний шар над безводним сульфатом натрію, фільтрують, та концентрують під зниженим тиском, одержуючи вказану в заголовку сполуку (160 г, 91 %) у вигляді в'язкого + масла. ES-MS m/z 246,1 [M+1] . Підготовчий синтез 25 Гідрохлорид (2R, 4R)-4-гідроксипіролідин-2-карбонової кислоти До суміші оцтового ангідриду (408 г) та оцтової кислоти (1,2 л) додають однією порцією при 50C транс-4-гідрокси-L-пролін (0,36 моль, 94 г). Нагрівають реакційну суміш до 90C, витримують при цій температурі протягом 5,5 год., після чого її концентрують. Розчиняють залишок у 2 н хлористоводневій кислоті, та нагрівають одержану суміш зі зворотним холодильником протягом 3 год. Охолоджують реакційну суміш до кімнатної температури, фільтрують через діатомову землю, та концентрують у вакуумі до утворення білих голчастих кристалів. Кристали відділяють фільтруванням, промивають діетиловим ефіром, та сушать у 20 вакуумі, одержуючи вказану в заголовку сполуку (90,0 г, 75 %). []D +10,0 (c=1,0 у метанолі). 1 H ЯМР (400 МГц, D2O), 2,34-2,39 (m, 1H), 2,45-2,53 (m, 1H), 3,38 (dd, 1H), 3,45 (d, 1H), 4,50 (dd, 1H), 4,58 (br s, 1H). Підготовчий синтез 26 2-метиловий складний ефір 1-трет-бутилового складного ефіру (2R, 4R)-4-гідроксипіролідин 26 UA 100131 C2 5 10 15 20 25 30 35 40 45 50 55 60 1,2-дикарбонової кислоти Додають краплями тіонілхлорид (1,81 моль, 213,8 г, 1,5 екв.) до розчину гідрохлориду (2R, 4R)-4-гідроксипіролідин-2-карбонової кислоти (1,19 моль, 200 г) у безводному метанолі (2 л) при 0C в атмосфері азоту. Після закінчення додавання нагрівають реакційну суміш до кімнатної температури, та перемішують протягом 6 год. Концентрують реакційну суміш під зниженим тиском, і одержують гідрохлорид відповідного метилового складного ефіру. До розчину гідрохлориду метилового складного ефіру у безводному дихлорметані (2 л) при 0C додають триетиламін (265,9 г, 2,63 моль), та перемішують протягом 30 хв. Потім додають послідовно N, N-диметиламінопіридин (0,18 моль, 21,9 г) та ди-трет-бутилдикарбонат (1,43 моль, 313,5 г). підігрівають реакційну суміш до кімнатної температури, та перемішують протягом 18 год. Гасять реакційну суміш водою, відділяють органічний шар, та промивають його водою та розчином NaHCO3. Органічний шар сушать над безводним сульфатом натрію, фільтрують, та концентрують у вакуумі, одержуючи вказану в заголовку сполуку (260 г, 88 %) у вигляді густого 1 масла. H ЯМР (400 МГц, CDCl3) 1,43 (s, 9H), 1,46 (s, 9H), 2,05-2,10 (m, 2H), 2,26-2,35 (m, 2H), 3,48-3,56 (m, 2H), 3,58-3,61 (m, 1H), 3,64-3,70 (m, 2H), 3,77 (s, 3H), 3,79 (s, 3H), 4,27-4,29 (m, 1H), 4,34-4,38 (m, 2H). Підготовчий синтез 27 2-метиловий складний ефір 1-трет-бутилового складного ефіру (2S, 4R)-4метансульфонілоксипіролідин-1,2-дикарбонової кислоти Додають краплями метансульфонілхлорид (1,56 моль, 179 г) до розчину 2-метилового складного ефіру 1-трет-бутилового складного ефіру (2S, 4R)-4-гідроксипіролідин-1,2дикарбонової кислоти (0,65 моль, 160 г) у піридині (600 мл) при 0C в атмосфері азоту. Після закінчення додавання нагрівають реакційну суміш до кімнатної температури, та перемішують протягом 2 год. Розводять реакційну суміш дихлорметаном, промивають 0,1 н хлористоводневою кислотою та насиченим розчином бікарбонату натрію. Сушать органічний шар над безводним сульфатом натрію, фільтрують, та концентрують у вакуумі, одержуючи + вказану в заголовку сполуку (185 г, 91 %) у вигляді густого масла. ES-MS m/z 324,1 [M+1] , 224,0 + [M-Boc] . Підготовчий синтез 28 2-метиловий складний ефір 1-трет-бутилового складного ефіру (2R, 4R)-4метансульфонілоксипіролідин-1,2-дикарбонової кислоти Вказану в заголовку сполуку одержують за методикою, описаною у Підготовчому синтезі 27, використовуючи як вихідну сполуку 2-метиловий складний ефір 1-трет-бутилового складного + ефіру (2R, 4R)-4-гідроксипіролідин-1,2-дикарбонової кислоти. ES-MS m/z 324,1 [M+1] . Підготовчий синтез 29 2-метиловий складний ефір 1-трет-бутилового складного ефіру (2S, 4S)-4-бромпіролідин1,2-дикарбонової кислоти Додають частинами трифенілфосфін (0,88 моль, 231 г) до розчину 2-метилового складного ефіру 1-трет-бутилового складного ефіру (2S, 4R)-4-гідроксипіролідин-1,2-дикарбонової кислоти (0,58 моль, 144 г) та CBr4 (0,88 моль, 292 г) у безводному дихлорметані (1,44 л) при 0C. Після закінчення додавання нагрівають реакційну суміш до кімнатної температури, та перемішують протягом 4 год. Додають етанол (1,44 л), та перемішують ще протягом 2 год. Додають до реакційної суміші діетиловий ефір (1,44 л), відфільтровують трифенілфосфіноксид, який випадає в осад, та концентрують фільтрат під зниженим тиском. Очищають продукт хроматографією на колонці, використовуючи як елюент 5 % етилацетат в гексані, і одержують + вказану в заголовку сполуку (155 г, 85 %) у вигляді густого масла. ES-MS m/z 308 [M+1] . Підготовчий синтез 30 2-метиловий складний ефір 1-трет-бутилового складного ефіру (2S, 4S)-4-бром-піролідин1,2-дикарбонової кислоти Вказану в заголовку сполуку одержують за методикою, описаною у Підготовчому синтезі 29, використовуючи як вихідну сполуку 2-метиловий складний ефір 1-трет-бутилового складного 1 ефіру (2R, 4R)-4-гідроксипіролідин-1,2-дикарбонової кислоти. H ЯМР (400 МГц, CDCl3) 1,451,47 (s, 9H), 2,38-2,45 (m, 1H), 2,54-2,59 (m, 1H), 3,72-3,74 (s, 3H), 3,84-3,87 (m, 1H), 3,92-3,94 (m, 1H), 4,44-4,54 (m, 2H). Підготовчий синтез 31 2-метиловий складний ефір 1-трет-бутилового складного ефіру (2S, 4S)-4-азидопіролідин1,2-дикарбонової кислоти Нагрівають суміш 2-метилового складного ефіру 1-трет-бутилового складного ефіру (2S, 4R)-4-метансульфонілоксипіролідин-1,2-дикарбонової кислоти (0,60 моль, 185 г) та азиду натрію (1,20 моль, 78 г) у N, N-диметилформаміді (1 л) до 80C, і витримують згадану суміш при цій 27 UA 100131 C2 5 температурі протягом 16 год. Розводять реакційну суміш водою, та екстрагують етилацетатом. Промивають об'єднані органічні шари 0,5н хлористоводневою кислотою, насиченим розчином бікарбонату натрію та розсолом. Сушать над безводним сульфатом натрію, фільтують, та концентрують у вакуумі, одержуючи вказану в заголовку сполуку (140 г, 88 %) у вигляді густого + + жовтого масла. ES-MS m/z 271,2 [M+1] , 171,0 [M-Boc] . Проміжні продукти, перелічені у поданій нижче таблиці, одержують загалом за методикою, описаною у Підготовчому синтезі 31, використовуючи відповідні бром- або мезилпіролідини. Підг. синт. * 32 33 34 Хімічна назва 2-метиловий складний ефір 1трет-бутилового складного ефіру (2S, 4R)-4азидопіролідин-1,2дикарбонової кислоти 2-метиловий складний ефір 1трет-бутилового складного ефіру (2R, 4R)-4азидопіролідин-1,2дикарбонової кислоти 2-метиловий складний ефір 1трет-бутилового складного ефіру (2R, 4S)-4азидопіролідин-1,2дикарбонової кислоти Фізичні дані H ЯМР (400 МГц, CDCl3) 1,40-1,41 (s, 9H), 2,092,13 (m, 1H), 2,40-2,44 (m, 1H), 3,37-3,45 (m, 1H), 3,61-3,65 (m, 1H), 3,67-3,69 (s, 3H), 4,09-4,15 (m, 1H), 4,25-4,38 (m, 1H) 1 + + ES-MS m/z 271 [M+1] , 171,1 [M-Boc] основний пік * Витримують при 65C протягом 16 год 10 15 20 25 Підготовчий синтез 35 Трет-бутиловий складний ефір (2S, 4S)-4-азидо-2-гідроксиметилпіролідин-1-карбонової кислоти Додають боргідрид літію (0,16 моль, 3,6 г) до розчину 2-метилового складного ефіру 1-третбутилового складного ефіру (2S, 4S)-4-азидопіролідин-1,2-дикарбонової кислоти (0,16 моль, 40 г) у безводному діетиловому ефірі (400 мл) при температурі від -10C до -20C в атмосфері азоту. Після закінчення додавання перемішують реакційну суміш протягом 30 хв при тій самій температурі. Гасять реакційну суміш насиченим розчином бікарбонату натрію при -70C, та дозволяють повільно нагрітися до кімнатної температури. Відділяють органічний шар, та екстрагують водний шар дихлорметаном. Сушать органічний шар над безводним сульфатом натрію, фільтрують, та концентрують у вакуумі, одержуючи вказану в заголовку сполуку (32 г, 1 неочищену) у вигляді густого жовтого масла. H ЯМР (400 МГц, CDCl3) 1,46 (s, 9H), 2,32-2,35 (m, 1H), 3,30 (dd, 1H), 3,65-3,79 (m, 4H), 4,07-4,29 (m, 2H). Проміжні продукти, перелічені у поданій нижче таблиці, одержують загалом за методикою, описаною у Підготовчому синтезі 35, використовуючи як вихідні сполуки відповідні метилові складні ефіри піролідин-2-карбонової кислоти. Підгот. синтез 36 37 38 30 Хімічна назва Трет-бутиловий складний карбонової кислоти Трет-бутиловий складний карбонової кислоти Трет-бутиловий складний карбонової кислоти ефір (2S, 4R)-4-азидо-2-гідроксиметилпіролідин-1 ефір (2R, 4R)-4-азидо-2-гідроксиметилпіролідин-1 ефір (2R, 4S)-4-азидо-2-гідроксиметилпіролідин-1 Підготовчий синтез 39 Трет-бутиловий складний ефір (2S, 4S)-4-аміно-2-гідроксиметилпіролідин-1-карбонової кислоти 28
ДивитисяДодаткова інформація
Назва патенту англійською6h-dibenzo[b,e]oxepine derived nonsteroidal mineralocorticoid receptor antagonists
Автори англійськоюGavardinas, Konstantinos, Jadhav, Prabhakar, Kondaji
Назва патенту російськоюПроизводные 6н-дибензо[b,e]оксепина как нестероидные антагонисты рецепторов минералокортикоидов
Автори російськоюГавардинас Константинос, Джадхав Прабхакар Кондаи
МПК / Мітки
МПК: A61P 5/40, C07D 498/04, C07D 405/06, A61K 31/4184, C07D 405/14
Мітки: нестероїдні, 6н-дибензо[b,e]оксепіну, рецепторів, похідні, мінералокортикоїдів, антагоністи
Код посилання
<a href="https://ua.patents.su/59-100131-pokhidni-6n-dibenzobeoksepinu-yak-nesterodni-antagonisti-receptoriv-mineralokortikodiv.html" target="_blank" rel="follow" title="База патентів України">Похідні 6н-дибензо[b,e]оксепіну як нестероїдні антагоністи рецепторів мінералокортикоїдів</a>
Азабіциклічні, азатрициклічні і азаспіроциклічні похідні аміноциклогексану як антагоністи рецепторів nmda, 5ht і нейронних нікотинових рецепторів

Номер патенту: 77778
Опубліковано: 15.01.2007
Автори: Йіргенсонс Айгарс, Парсонс Крістофер Грахам Рафаель, Каусс Валерьянс, Ванейєвс Максімс, Даниш Войцех, Хенріх Маркус, Гольд Маркус, Калвіньш Іварс
МПК: A61P 25/02, A61P 25/28, A61P 25/22, A61P 37/02, A61P 21/00, C07D 209/54, A61P 25/06, A61P 27/06, C07D 487/08, A61P 25/08, A61P 33/06, A61P 25/34, A61K 31/403, C07D 221/22, C07D 471/08, A61K 31/438, A61P 25/04, A61P 25/14, A61P 25/24, A61P 21/02, A61P 25/30, A61P 25/32, A61P 9/00, C07D 221/20, A61K 31/437, A61P 25/36, A61P 25/16, A61P 31/12, A61P 25/18, A61P 25/00, A61P 9/10, A61P 43/00, C07D 209/02, A61K 31/439, A61P 1/08, A61P 27/16, A61P 31/18, A61P 1/04, A61P 31/14
Мітки: похідні, аміноциклогексану, нікотинових, nmda, антагоністи, нейронних, азатрициклічні, рецепторів, азабіциклічні, азаспіроциклічні
Формула / Реферат:
1. Сполуки формули (1),де R і R1-R5, кожний незалежно, вибраний з С1-6-алкільних груп, С2-6-алкенільних груп, С2-6-алкінільних груп, С6-12-арил-С1-4-алкільних груп, необов'язково заміщених С6-12-арильних груп і у випадку R і R2-R5 - з атомів водню, за умови, що щонайменше один з R2 і R3 і щонайменше один з R4 і R5 не є воднем, або R і R1 разом являють собою...
Похідні триазолу як антагоністи рецепторів тахікінінів

Номер патенту: 79113
Опубліковано: 25.05.2007
Автори: Джангхейм Луіс Ніколаус, Ремік Дейвід Майкл, Робертсон Майкл Алан, Савін Кеннет Аллен, Мехл Брайан Стефан, Хембре Ерік Джеймз, Гардінієр Кевін Мет'ю, Амегадзі Алберт Кудзові, Гонг Жан Ерік
МПК: A61K 31/4196, A61K 31/4192, C07D 403/04, A61P 43/00, A61K 31/506, A61K 31/497, A61K 31/444, C07D 413/04, A61P 25/04, A61K 31/541, C07D 403/14, A61P 25/18, A61K 31/5025, A61P 11/06, A61K 31/437, A61P 25/00, C07D 401/04, A61P 1/04, A61K 31/4439, C07D 417/14, C07D 487/04, C07D 249/08, A61K 31/422, A61P 17/00, C07D 413/14, A61P 25/24, A61K 31/501, A61P 1/08, C07D 249/06, A61P 25/22, C07D 401/14, C07D 498/04, A61K 31/5377, A61K 31/4985
Мітки: рецепторів, тахікінінів, антагоністи, триазолу, похідні
Формула / Реферат:
1. Сполука Формули І:,де:D - С1-С3-алкандіїл;R1 - феніл,факультативно заміщений замісниками в кількості від одного до трьох, незалежно один від одного вибраними з групи, до якої входять галоген, С1-С4-алкіл, С1-С4-алкокси-, ціаногрупа, дифторметил, трифторметил та трифторметоксигрупа;R4 - радикал, вибраний з групи, до якої...
Похідні імідазолу як антагоністи глутаматних рецепторів

Номер патенту: 80888
Опубліковано: 12.11.2007
Автори: Віера Ерік, Портер Річард Х'ю Філліп, Колчевскі Сабіне, Сессареллі Сімона Маріа, Буеттелманн Бернд, Ешке Георг
МПК: A61P 25/28, C07D 401/14, C07D 401/06, A61K 31/4439, A61K 31/444
Мітки: рецепторів, антагоністи, похідні, глутаматних, імідазолу
Формула / Реферат:
1. Сполука загальної формули, (I)де R1 означає галоген або ціано;R2 означає С1-С6-алкіл;R3 означає феніл або 5- або 6-членний гетероарил, який необов'язково заміщений одним, двома або трьома замісниками, вибраними з групи, що містить галоген, С1-С6-алкіл, С3-С6-циклоалкіл, С1-С6-алкілгалоген, ціано, С1-С6-алкокси, NR'R",...
Похідні карбоксаміду як антагоністи мускаринових рецепторів

Номер патенту: 89846
Опубліковано: 10.03.2010
Автори: Ватсон Крістін Анн Луіс, Вуд Ентоні, Стренг Росс Сінклер, Глоссоп Пол Алан, Мантелл Саймон Джон
МПК: C07D 413/12, C07D 211/46, C07D 205/00, C07D 207/12, A61K 31/40, A61P 37/00, A61K 31/4409, A61K 31/397
Мітки: рецепторів, антагоністи, похідні, карбоксаміду, мускаринових
Формула / Реферат:
1. Сполука формули (І), (I)в якій,R1 є CN або CONH2;А вибирають з, або ,де * і ** представляють точки приєднання, ** є місцем...
Похідні сульфонілпіразол та сульфонілпіразолон карбоксамідину як антагоністи рецепторів 5-ht6

Номер патенту: 98122
Опубліковано: 25.04.2012
Автори: Кейзер Гіскіас Г., ван Лувезейн Арнольд, Крузе Корнеліс Г., Івема Баккер Воутер І., Зоргдрагер Ян, ван дер Ньот Мартіна А.В.
МПК: C07D 407/04, C07D 513/04, C07D 409/14, C07D 401/12, C07D 409/04, C07D 231/12, C07D 409/12, C07D 407/12, C07D 401/04, A61P 25/00, C07D 417/12, A61K 31/415, C07D 231/06, C07D 403/12, A61K 31/4155
Мітки: карбоксамідину, рецепторів, антагоністи, сульфонілпіразол, сульфонілпіразолон, похідні, 5-ht6
Формула / Реферат:
1. Сполука формули (1)або її таутомер, стереоізомер, N-оксид або фармакологічно прийнятна сіль, гідрат або сольват будь-яких з вищевказаних, де:R1 представляє водень, незаміщену алкіл(C1-4)групу, алкіл(C1-4)групу, заміщену одним або більше атомами галогену, R2 та R3 незалежно представляють водень, незаміщену алкіл(C1-4)групу, алкіл(C1-4)групу,...
Попередній патент: Нарізне з’єднання, що містить щонайменше один нарізний елемент із торцевою крайкою для металевої труби
Наступний патент: Похідні ізоксазолопіридину
Випадковий патент: Стиракрилова композиція для клейових з'єднань у будівництві