2-феніліндоли як антагоністи рецепторів простагландину d2
Номер патенту: 95303
Опубліковано: 25.07.2011
Автори: Метью Роуз М., Харріс Кіт Дж., Ян Чжаося, Райлінг Штефан, Джексон Шерон, Недузак Таддеуш Р.





























Формула / Реферат
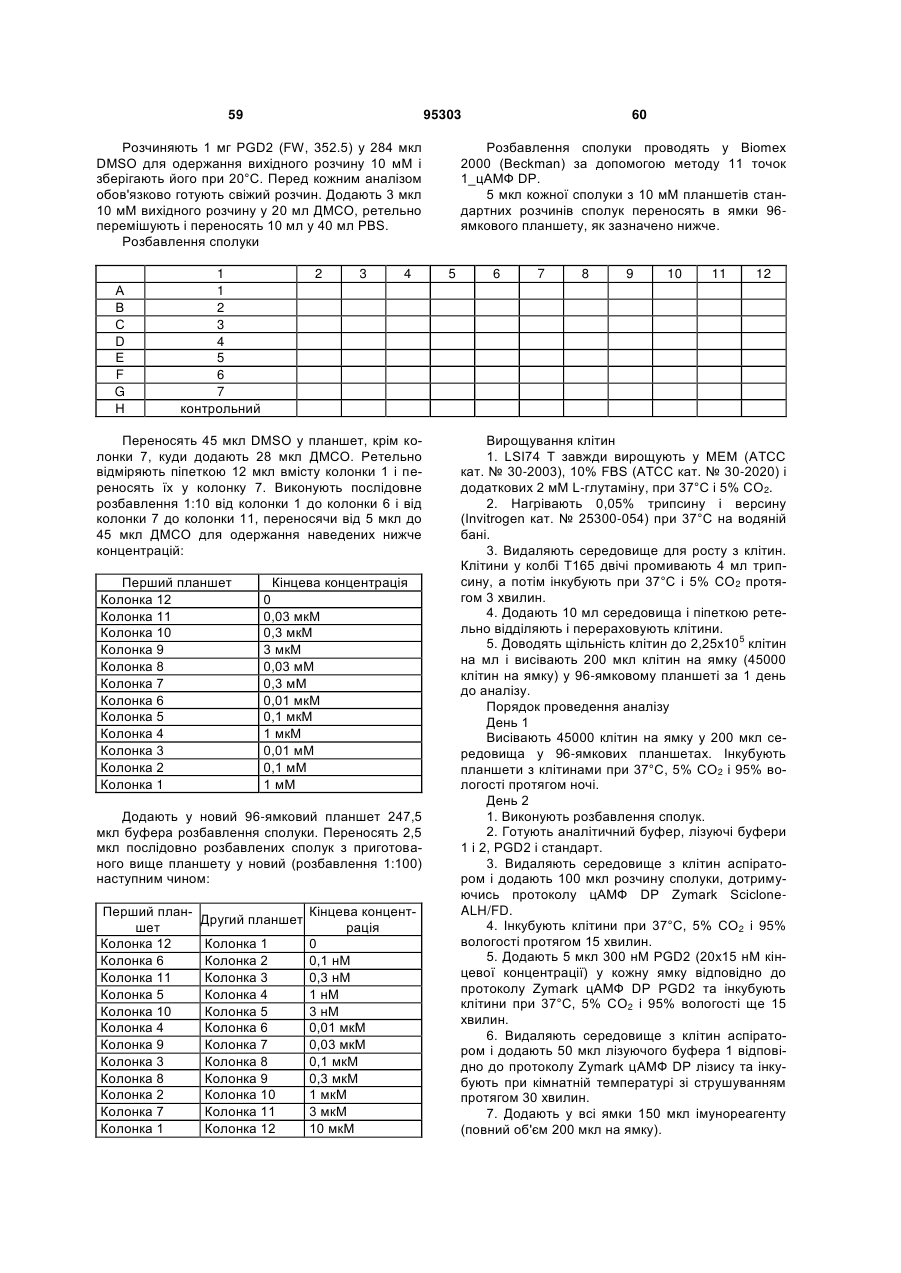
1. Сполука формули (А):
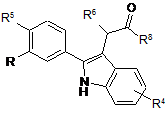 , (А)
, (А)
де
R являє собою R1CH2SO2-, R2CH2SO2NH- або R3NHSO2-,
R1 являє собою феніл, необов'язково заміщений галогеном,
R2 являє собою феніл, заміщений галогеном,
R3 являє собою 2,6-дихлорбензил, 3,5-дихлорбензил, 2,4-дихлорфенілетил, 2-метоксифенілетил, 3-метоксифенілетил, 4-метоксифенілетил, 2-трифторметилфенілетил, феніл етил або 3-феніл-н-пропіл,
R4 являє собою водень,
R5 являє собою хлор,
R6 являє собою водень і
R8 являє собою гідрокси; або
R являє собою циклогексиламіносульфоніл,
R4 являє собою 4-хлор, 4-фтор, 4-метил або 7-хлор,
R5 являє собою хлор або етил,
R6 являє собою водень або метил і
R8 являє собою гідрокси; або
R являє собою циклогексиламіносульфоніл,
R4 являє собою водень,
R5 являє собою хлор,
R6 являє собою водень,
R8 являє собою -NHR7 і
R7 являє собою метил, метилсульфоніл, етилсульфоніл, галогеналкілсульфоніл або тетразоліл;
або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків.
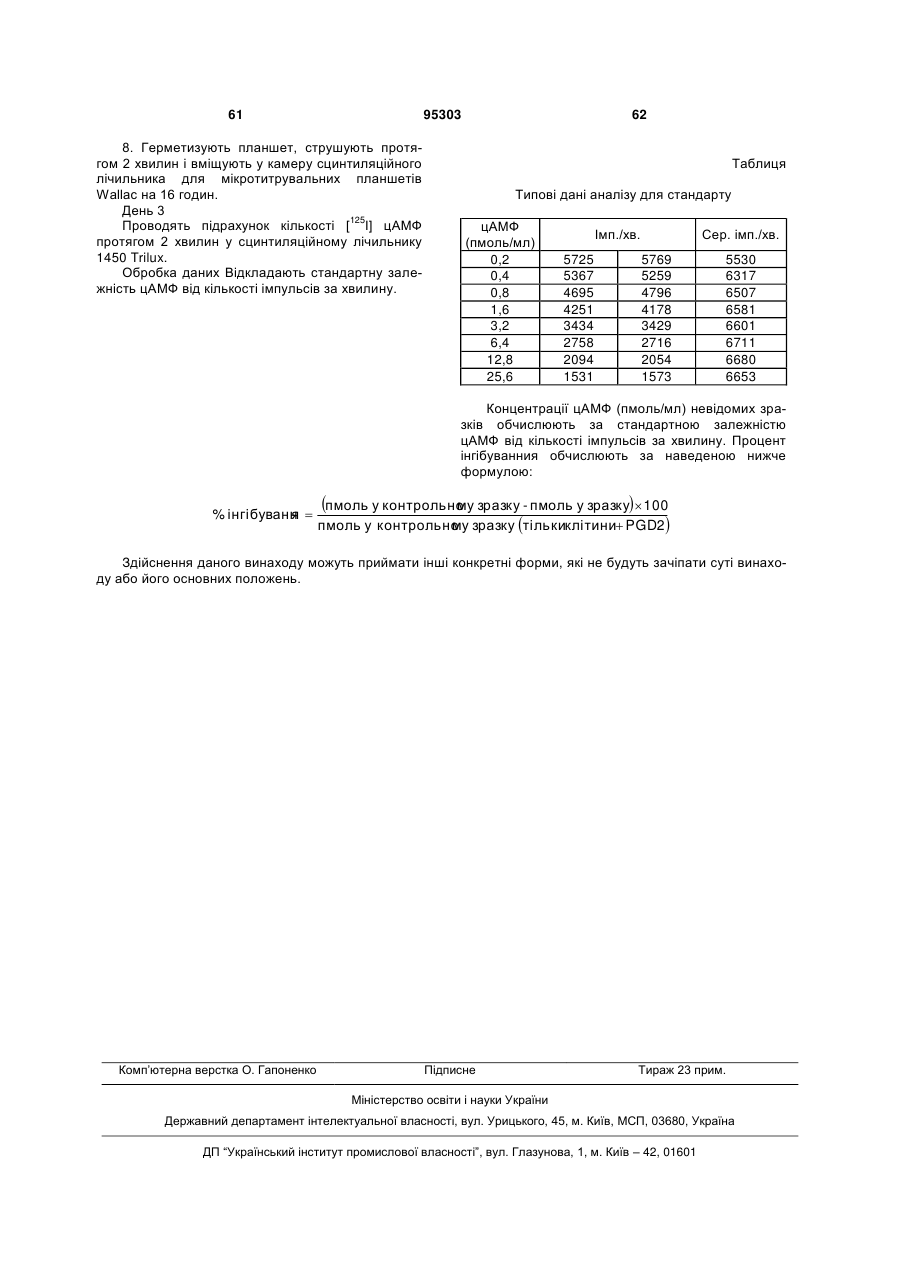
2. Сполука за п. 1, яка є сполукою формули (І):
 , (I)
, (I)
де
R являє собою R1CH2SO2-, R2CH2SO2NH- або R3NHSO2-,
R1 являє собою феніл, необов'язково заміщений галогеном,
R2 являє собою феніл, заміщений галогеном, і
R3 являє собою 2,6-дихлорбензил, 3,5-дихлорбензил, 2,4-дихлорфенілетил, 2-метоксифенілетил, 3-метоксифенілетил, 4-метоксифенілетил, 2-трифторметилфенілетил, фенілетил або 3-феніл-н-пропіл;
або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків.
3. Сполука за п. 2, в якій R являє собою R3NHSO2-, або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків.
4. Сполука за п. 1, яка є сполукою формули (II):
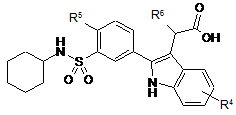 , (ІІ)
, (ІІ)
де
R4 являє собою 4-хлор, 4-фтор, 4-метил або 7-хлор,
R5 являє собою хлор або етил і
R6 являє собою водень або метил,
або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків.
5. Сполука за п. 4, в якій R5 являє собою хлор і R6 являє собою водень, або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків.
6. Сполука за п. 1, яка є сполукою формули (III):
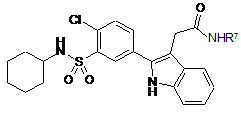 , (III)
, (III)
де
R7 являє собою метил, метилсульфоніл, етилсульфоніл, галогеналкілсульфоніл або тетразоліл,
або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків.
7. Сполука за п. 1, що вибрана з групи:
{2-[4-хлор-3-(2,6-дихлорбензилсульфамоїл)феніл]-1H-індол-3-іл}оцтової кислоти,
{2-[4-хлор-3-(3,5-дихлорбензилсульфамоїл)феніл]-1H-індол-3-іл}оцтової кислоти,
(2-{4-хлор-3-[2-(2,4-дихлорфеніл)етилсульфамоїл]феніл}-1H-індол-3-іл)оцтової кислоти,
(2-{4-хлор-3-[2-(2-метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3-іл)оцтової кислоти,
(2-{4-хлор-3-[2-(3-метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3-іл)оцтової кислоти,
(2-{4-хлор-3-[2-(4-метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3-іл)оцтової кислоти,
(2-{4-хлор-3-[2-(2-трифторметоксифеніл)етилсульфамоїл]феніл}-1H-індол-3-іл)оцтової кислоти,
[2-(4-хлор-3-фенетилсульфамоїлфеніл)-1Н-індол-3-іл]оцтової кислоти,
{2-[4-хлор-3-(3-фенілпропілсульфамоїл)феніл]-1Н-індол-3-іл}оцтової кислоти,
{2-[4-хлор-3-(3-хлорфенілметансульфоніл)феніл]-1H-індол-3-іл}оцтової кислоти,
{2-[4-хлор-3-(3-хлорфенілметансульфоніламіно)феніл]-1Н-індол-3-іл}оцтової кислоти,
[4-хлор-2-(4-хлор-3-циклогексилсульфамоїлфеніл)-1Н-індол-3-іл]оцтової кислоти,
[2-(4-хлор-3-циклогексилсульфамоїлфеніл)-4-фтор-1H-індол-3-іл]оцтової кислоти,
[2-(4-хлор-3-циклогексилсульфамоїлфеніл)-4-метил-1H-індол-3-іл]оцтової кислоти,
[7-хлор-2-(4-хлор-3-циклогексилсульфамоїлфеніл)-1H-індол-3-іл]оцтової кислоти,
[2-(3-циклогексилсульфамоїл-4-етилфеніл)-1Н-індол-3-іл]оцтової кислоти,
2-[2-(4-хлор-3-циклогексилсульфамоїлфеніл)-1Н-індол-3-іл]пропіонової кислоти,
2-[2-(4-хлор-3-циклогексилсульфамоїлфеніл)-1Н-індол-3-іл]-N-метилацетаміду,
2-хлор-N-циклогексил-5-[3-(2-метансульфоніламіно-2-оксоетил)-1Н-індол-2-іл]бензолсульфаміду,
2-хлор-N-циклогексил-5-[3-(2-етансульфоніламіно-2-оксоетил)-1Н-індол-2-іл]бензолсульфаміду,
2-хлор-N-циклогексил-5-[3-(2-оксо-2-трифторметансульфоніламіноетил)-1Н-індол-2-іл]бензолсульфаміду або
2-[2-(4-хлор-3-циклогексилсульфамоїлфеніл)-1Н-індол-3-іл]-N-(1Н-тетразол-5-іл)ацетаміду,
або її фармацевтично прийнятної солі, гідрату або сольвату, її фармацевтично прийнятних проліків або фармацевтично прийнятної солі, гідрату або сольвату проліків.
8. Фармацевтично прийнятна сіль сполуки за п. 1, що являє собою [4-хлор-2-(4-хлор-3-циклогексилсульфамоїлфеніл)-1H-індол-3-іл]ацетат калію.
9. Фармацевтична композиція, що містить фармацевтично ефективну кількість сполуки за п. 1 або її фармацевтично прийнятної солі, гідрату або сольвату, її фармацевтично прийнятних проліків або фармацевтично прийнятної солі, гідрату або сольвату проліків у суміші з фармацевтично прийнятним носієм.
10. Спосіб лікування алергічного захворювання, системного мастоцитозу, порушення, що супроводжується системною активацією мастоцитів, анафілактичного шоку, бронхоконстрикції, бронхіту, екземи, захворювань, що супроводжуються свербежем, захворювань, які виникають як вторинні захворювання внаслідок поведінки, що супроводжується свербежем, хронічних обструктивних захворювань легенів, ішемічного реперфузійного ушкодження, розладу мозкового кровообігу, хронічного ревматоїдного артриту, плевриту або виразкового коліту у пацієнта, який потребує такого лікування, при якому вводять такому пацієнту фармацевтично ефективну кількість сполуки за п. 1 або її фармацевтично прийнятної солі, гідрату або сольвату, її фармацевтично прийнятних проліків або фармацевтично прийнятної солі, гідрату або сольвату проліків.
11. Спосіб за п. 10, в якому поведінкою, що супроводжується свербежем, є розчухування або розтирання.
12. Спосіб за п. 10, в якому захворюванням, що виникає як вторинне захворювання внаслідок поведінки, що супроводжується свербежем, є катаракта, відшаровування сітківки, запалення, інфекція або порушення сну.
13. Спосіб за п. 10, в якому алергічним захворюванням є алергічний риніт, алергічний кон'юнктивіт, атопічний дерматит, бронхіальна астма або харчова алергія.
14. Спосіб за п. 10, в якому захворюванням, що супроводжується свербежем, є атопічний дерматит або кропивниця.
15. Фармацевтична композиція, що містить фармацевтично ефективну кількість сполуки за п. 1 та сполуку, вибрану з групи, що включає антигістамін, антагоніст лейкотриєну, бета-антагоніст, інгібітор PDE4, антагоніст ТР і CrTh2, у суміші з фармацевтично прийнятним носієм.
16. Фармацевтична композиція за п. 15, де антигістаміном є фексофенадин, лоратадин, деслоратадин або цетиризин, антагоністом лейкотриєну є монтелукаст або зафірлукаст, бета-антагоністом є альбутерол, сальбутерол або тербуталін, інгібітором PDE4 є рофлуміласт або циломіласт, антагоністом ТР є раматробан і антагоністом CrTh2 є раматробан.
Текст
1. Сполука формули (А): O R6 R5 2 3 95303 фармацевтично прийнятна сіль, гідрат або сольват проліків. 3 3. Сполука за п. 2, в якій R являє собою R NHSO2-, або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків. 4. Сполука за п. 1, яка є сполукою формули (II): O R6 R5 OH H N S O O N H R4 , (ІІ) де 4 R являє собою 4-хлор, 4-фтор, 4-метил або 7хлор, 5 R являє собою хлор або етил і 6 R являє собою водень або метил, або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків. 5 6 5. Сполука за п. 4, в якій R являє собою хлор і R являє собою водень, або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків. 6. Сполука за п. 1, яка є сполукою формули (III): O Cl NHR7 H N S O O N H , (III) де 7 R являє собою метил, метилсульфоніл, етилсульфоніл, галогеналкілсульфоніл або тетразоліл, або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків. 7. Сполука за п. 1, що вибрана з групи: {2-[4-хлор-3-(2,6-дихлорбензилсульфамоїл)феніл]1H-індол-3-іл}оцтової кислоти, {2-[4-хлор-3-(3,5-дихлорбензилсульфамоїл)феніл]1H-індол-3-іл}оцтової кислоти, (2-{4-хлор-3-[2-(2,4дихлорфеніл)етилсульфамоїл]феніл}-1H-індол-3іл)оцтової кислоти, (2-{4-хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти, (2-{4-хлор-3-[2-(3метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти, (2-{4-хлор-3-[2-(4метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти, 4 (2-{4-хлор-3-[2-(2трифторметоксифеніл)етилсульфамоїл]феніл}-1Hіндол-3-іл)оцтової кислоти, [2-(4-хлор-3-фенетилсульфамоїлфеніл)-1Н-індол3-іл]оцтової кислоти, {2-[4-хлор-3-(3-фенілпропілсульфамоїл)феніл]-1Ніндол-3-іл}оцтової кислоти, {2-[4-хлор-3-(3-хлорфенілметансульфоніл)феніл]1H-індол-3-іл}оцтової кислоти, {2-[4-хлор-3-(3хлорфенілметансульфоніламіно)феніл]-1Н-індол3-іл}оцтової кислоти, [4-хлор-2-(4-хлор-3-циклогексилсульфамоїлфеніл)1Н-індол-3-іл]оцтової кислоти, [2-(4-хлор-3-циклогексилсульфамоїлфеніл)-4фтор-1H-індол-3-іл]оцтової кислоти, [2-(4-хлор-3-циклогексилсульфамоїлфеніл)-4метил-1H-індол-3-іл]оцтової кислоти, [7-хлор-2-(4-хлор-3-циклогексилсульфамоїлфеніл)1H-індол-3-іл]оцтової кислоти, [2-(3-циклогексилсульфамоїл-4-етилфеніл)-1Ніндол-3-іл]оцтової кислоти, 2-[2-(4-хлор-3-циклогексилсульфамоїлфеніл)-1Ніндол-3-іл]пропіонової кислоти, 2-[2-(4-хлор-3-циклогексилсульфамоїлфеніл)-1Ніндол-3-іл]-N-метилацетаміду, 2-хлор-N-циклогексил-5-[3-(2метансульфоніламіно-2-оксоетил)-1Н-індол-2іл]бензолсульфаміду, 2-хлор-N-циклогексил-5-[3-(2-етансульфоніламіно2-оксоетил)-1Н-індол-2-іл]бензолсульфаміду, 2-хлор-N-циклогексил-5-[3-(2-оксо-2трифторметансульфоніламіноетил)-1Н-індол-2іл]бензолсульфаміду або 2-[2-(4-хлор-3-циклогексилсульфамоїлфеніл)-1Ніндол-3-іл]-N-(1Н-тетразол-5-іл)ацетаміду, або її фармацевтично прийнятної солі, гідрату або сольвату, її фармацевтично прийнятних проліків або фармацевтично прийнятної солі, гідрату або сольвату проліків. 8. Фармацевтично прийнятна сіль сполуки за п. 1, що являє собою [4-хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1H-індол-3іл]ацетат калію. 9. Фармацевтична композиція, що містить фармацевтично ефективну кількість сполуки за п. 1 або її фармацевтично прийнятної солі, гідрату або сольвату, її фармацевтично прийнятних проліків або фармацевтично прийнятної солі, гідрату або сольвату проліків у суміші з фармацевтично прийнятним носієм. 10. Спосіб лікування алергічного захворювання, системного мастоцитозу, порушення, що супроводжується системною активацією мастоцитів, анафілактичного шоку, бронхоконстрикції, бронхіту, екземи, захворювань, що супроводжуються свербежем, захворювань, які виникають як вторинні захворювання внаслідок поведінки, що супроводжується свербежем, хронічних обструктивних захворювань легенів, ішемічного реперфузійного ушкодження, розладу мозкового кровообігу, хронічного ревматоїдного артриту, плевриту або виразкового коліту у пацієнта, який потребує такого лікування, при якому вводять такому пацієнту фармацевтично ефективну кількість сполуки за п. 5 95303 6 1 або її фармацевтично прийнятної солі, гідрату або сольвату, її фармацевтично прийнятних проліків або фармацевтично прийнятної солі, гідрату або сольвату проліків. 11. Спосіб за п. 10, в якому поведінкою, що супроводжується свербежем, є розчухування або розтирання. 12. Спосіб за п. 10, в якому захворюванням, що виникає як вторинне захворювання внаслідокповедінки, що супроводжується свербежем, є катаракта, відшаровування сітківки, запалення, інфекція або порушення сну. 13. Спосіб за п. 10, в якому алергічним захворюванням є алергічний риніт, алергічний кон'юнктивіт, атопічний дерматит, бронхіальна астма або харчова алергія. 14. Спосіб за п. 10, в якому захворюванням, що супроводжується свербежем, є атопічний дерматит або кропивниця. 15. Фармацевтична композиція, що містить фармацевтично ефективну кількість сполуки за п. 1 та сполуку, вибрану з групи, що включає антигістамін, антагоніст лейкотриєну, бета-антагоніст, інгібітор PDE4, антагоніст ТР і CrTh2, у суміші з фармацевтично прийнятним носієм. 16. Фармацевтична композиція за п. 15, де антигістаміном є фексофенадин, лоратадин, деслоратадин або цетиризин, антагоністом лейкотриєну є монтелукаст або зафірлукаст, бета-антагоністом є альбутерол, сальбутерол або тербуталін, інгібітором PDE4 є рофлуміласт або циломіласт, антагоністом ТР є раматробан і антагоністом CrTh2 є раматробан. Галузь винаходу Даний винахід стосується 2-феніліндольних сполук, їх одержання, фармацевтичних композицій, що містять ці сполуки, і їх фармацевтичного використання у лікуванні патологічних станів, які можна контролювати інгібуванням рецептора простагландину D2. Рівень техніки Показано, що локальна стимуляція алергеном пацієнтів з алергічним ринітом, бронхіальною астмою, алергічним кон'юнктивітом і атопічним дерматитом приводить до швидкого зростання рівня простагландину D2 «(PGD2)» у назальній і бронхіальній змивній рідини, сльозах і рідині, зібраній зі шкірного міхура за методом «шкірної камери». PGD2 може здійснювати ряд запальних дій, наприклад підвищувати проникність судин у кон'юнктиві і шкірі, підвищувати опір дихальних шляхів носа, звуження дихальних шляхів і проникнення еозинофілів у кон'юнктиву і трахеї. PGD2 - основний циклооксигеназний продукт арахідонової кислоти, що продукується мастоцитами при імунологічному стимулюванні [Lewis, RA, Soter ΝΑ, Diamond PT, Austen KF, Oates JA, Roberts LJ II, prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE, J. Immunol. 129, 1627-1631, 1982]. Активовані мастоцити, одне з головних джерел PGD2, грають одну з ключових ролей у виникненні алергічної реакції при таких захворюваннях як астма, алергічний риніт, алергічний кон'юнктивіт, алергічний дерматит та інші захворювання [Brightling СЕ, Bradding Ρ, Pavord ID, Wardlaw AJ, New Insights into the role of the mast cell in asthma, Clin Exp Allergy 33, 550556, 2003]. Багато впливів PGD2 опосередковується його дією на рецептор простагландину D («DP») - рецептор, зв'язаний з G-білком, що експресується на епітелії та у гладких м'язах. При астмі респіраторний епітелій вже давно вважається головним джерелом запальних цитокінів і хемокінів, які сприяють прогресуванню захворювання [Holgate S, Lackie P, Wilson S, Roche W, Davies D, Bronchial Epithelium as a Key Regulator of Airway Allergen Sensitzation and Remodeling in Asthma, Am J Respir Crit Care Med. 162, 113-117, 2000]. В експериментальній моделі астми мишей при стимулюванні антигеном відбувається різка активація рецептора DP на епітелії дихальних шляхів [Matsuoka Τ, Hirata M, Tanaka Η, Takahashi Υ, Murata T, Kabashima Κ, Sugimoto Υ, Kobayashi Τ, Ushikubi F, Aze Υ, Eguchi Ν, Urade Υ, Yoshida Ν, Kimura K Mizoguchi A, Honda Y, Nagai H, Narumiya S, Prostaglandin D2 as a mediator of allergic asthma, Science 287, 2013-2017, 2000]. У нокаутних мишей з відсутнім рецептором DP помітно знижується гіперреактивність дихальних шляхів і хронічне запалення [Matsuoka Τ, Hirata M, Tanaka Η, Takahashi Υ, Murata T, Kabashima Κ, Sugimoto Υ, Kobayashi Τ, Ushikubi F, Aze Υ, Eguchi Ν, Urade Υ, Yoshida Ν, Kimura K, Mizoguchi A, Honda Y, Nagai H, Narumiya S, Prostaglandin D2 as a mediator of allergic asthma, Science 287, 2013-2017, 2000] - дві найважливіших ознаки астми людини. Вважається також, що рецептор DP задіяний в алергічному риніті людини, поширеному алергічному захворюванні, що характеризується такими симптомами як чихання, свербіж, ринорея і закладеність носа. Місцеве застосування PGD2 у носовій порожнині викликає залежне від дози збільшення закладеності носа [Doyle WJ, Boehm S, Skoner DP, Physiologic responses to intranasal doseresponse challenges with histamine, methacholine, bradykinin, and prostaglandin in adult volunteers with and without nasal allergy, J Allergy Clin Immunol. 86(6 Pt 1), 924-35, 1990]. Було показано, що антагоністи рецептора DP знижують запалення дихальних шляхів в експериментальній моделі астми морських свинок [Arimura A, Yasui K, Kishino J, Asanuma F, Hasegawa H, Kakudo S, Ohtani M, Arita Η (2001), Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S-5751, J. Pharmacol. Exp Ther. 298(2), 411-9, 2001]. Таким чином, PGD2, очевидно, діє на рецептор DP і грає важливу роль у виявленні основних особливостей алергічної астми. Було показано, що антагоністи DP можуть ефективно ослаблювати симптоми алергічного 7 риніту у багатьох видів, зокрема, було показано, що вони можуть придушувати індуковану антигенами закладеність носа, найбільш явний симптом алергічного риніту [Jones, T.R., Savoie, С, Robichaud, Α., Sturino, С, Scheigetz, J., Lachance, N.,Roy, В., Boyd, M., Abraham, W., Studies with a DP receptor antagonist in sheep and guinea pig models of allergic rhinitis, Am. J. Resp. Crit. Care Med. 167, A218, 2003; і Arimura A, Yasui K, Kishino J, Asanuma F, Hasegawa H, Kakudo S, Ohtani M, Arita H. Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S-5751. J. Pharmacol. Exp Ther. 298(2), 411-9, 2001]. Антагоністи DP також ефективні в експериментальних моделях алергічного кон'юнктивіту і алергічного дерматиту [Arimura A, Yasui K, Kishino J, Asanuma F, Hasegawa H, Kakudo S, Ohtani M, Arita H, Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S-5751. J. Pharmacol Exp Ther. 298(2), 411-9, 2001; і Torisu K, Kobayashi K, Iwahashi M, Nakai Y, Onoda T, Nagase T, Sugimoto I, Okada Y, Matsumoto R, Nanbu F, Ohuchida S, Nakai H, Toda M, Discovery of a new class of potent, selective, and orally active prostaglandin D2 receptor antagonists, Bioorg. & Med. Chem. 12, 5361-5378, 2004]. Короткий опис винаходу Даний винахід стосується сполуки формули (А): дe 1 2 R являє собою R CH2SO2-, R CH2SO2NH- або 3 R NHSO2-, 1 R являє собою феніл, необов'язково заміщений галогеном, 2 R являє собою феніл, заміщений галогеном, 3 R являє собою 2,6-дихлорбензил, 3,5дихлорбензил, 2,4-дихлорфенілетил, 2метоксифенілетил, 3-метоксифенілетил, 4метоксифенілетил, 2-трифторметил-фенілетил, фенілетил або 3-феніл-н-пропіл, 4 R являє собою водень, 5 R являє собою хлор, 6 R являє собою водень і 8 R являє собою гідрокси; або R являє собою циклогексиламіносульфоніл, 4 R являє собою 4-хлор, 4-фтор, 4-метил або 7хлор, 5 R являє собою хлор або етил, 6 R являє собою водень або метил, і 8 R являє собою гідрокси; або R являє собою циклогексиламіносульфоніл, 4 R являє собою водень, 5 R являє собою хлор, 6 R являє собою водень, 8 7 R являє собою -NHR і 95303 8 7 R являє собою метил, метилсульфоніл, етилсульфоніл, галогеналкіл-сульфоніл або тетразоліл; або її фармацевтично прийнятної солі, гідрату або сольвату, її фармацевтично прийнятних проліків або фармацевтично прийнятної солі, гідрату або сольвату проліків. Іншим аспектом даного винаходу є фармацевтична композиція, що включає фармацевтично ефективну кількість однієї або більше сполук винаходу або їх фармацевтично прийнятної солі, гідрату або сольвату, їх фармацевтично прийнятних проліків або фармацевтично прийнятної солі, гідрату або сольвату проліків у суміші з фармацевтично прийнятним носієм. Ще один аспект даного винаходу - спосіб лікування пацієнта, який страждає захворюванням, опосередкованим PGD2, у тому числі, зокрема, алергічним захворюванням (наприклад, алергічний риніт, алергічний кон'юнктивіт, атопічний дерматит, бронхіальна астма і харчова алергія), системним мастоцитозом, порушеннями, що супроводжуються системною активацією мастоцитів, анафілактичним шоком, бронхоконстрикцією, бронхітом, екземою, кропивницею, захворюваннями, що супроводжуються свербежем (наприклад, атопічний дерматит і кропивниця), захворюваннями (наприклад, катаракта, відшаровування сітківки, запалення, інфекція і порушення сну), які виникають як вторинні захворювання внаслідок поведінки, що супроводжується свербежем (наприклад, розчісування і розтирання), запаленням, хронічним обструктивним захворюванням легенів, ішемічним реперфузійним ушкодженням, розладом мозкового кровообігу, хронічним ревматоїдним артритом, плевритом, неспецифічним виразковим колітом і подібними захворюваннями, за допомогою введення такому пацієнту фармацевтично ефективної кількості сполуки винаходу або її фармацевтично прийнятної солі, гідрату або сольвату, її фармацевтично прийнятних проліків або фармацевтично прийнятної солі, гідрату або сольвату проліків. Докладний опис винаходу Визначення термінів Використовувані вище і у всьому тексті опису винаходу наведені нижче терміни, якщо не вказано інакше, мають зазначені далі прийняті значення: «Сполуки, що складають предмет даного винаходу» і аналогічні вирази включають описані вище у цьому документі сполуки формул (А), (І), (II) або (III), даний термін охоплює проліки, фармацевтично прийнятні солі і сольвати, наприклад, гідрати, де це допускається контекстом. Точно так само посилання на проміжні сполуки, незалежно від того, чи включені вони самі у формулу винаходу, покликане включати солі і сольвати у випадках, де це допускається контекстом. «Галогеналкіл» означає алкільну групу, що має від одного до трьох галогенів як замісники. У конкретному прикладі галогеналкілом є нижчі алкільні групи, що мають від одного до трьох галогенів як замісники. У більш конкретному випадку галогеналкілом є нижчі алкільні групи, заміщені одним галогеном. 9 «Галогеналкілсульфоніл» означає галогеналкіл-SО2-. Приклади включають CF3-SO2-. «Пацієнт» означає людину та інших ссавців. Використовуваний у цьому документі термін «фармацевтично прийнятні проліки» означає проліки сполук, що складають предмет даного винаходу, які у межах здорової медичної думки є придатними для використання у контакті з тканинами пацієнта, при цьому небажана токсичність, подразнення, алергічна реакція знаходяться у межах розумного співвідношення користь/ризик, а також є ефективними для застосування за призначенням сполук, що складають предмет даного винаходу. Термін «проліки» означає сполуку, яка in vivo трансформується з утворенням сполуки винаходу або її фармацевтично прийнятної солі, гідрату або сольвату. Трансформація може відбуватися за різними механізмами, наприклад за допомогою гідролізу у крові. Сполуки зі схильними до метаболічного розпаду групами мають ту перевагу, що вони можуть проявляти підвищену біодоступність внаслідок більш високої розчинності і/або швидкості всмоктування вихідної сполуки, завдяки наявності схильної до метаболічного розпаду групи, тому такі сполуки діють як проліки. Докладне обговорення наводиться у роботах Design of Prodrags, H. Bundgaard, ed., Elsevier (1985); Methods in Enzymology; K. Widder et al., Ed., Academic Press, 42, 309-396 (1985); A Textbook of Drug Design and Development, Krogsgaard-Larsen and H. Bundgaard, ed., Chapter 5; "Design and Applications of Prodrugs" 113-191 (1991); Advanced Drug Delivery Reviews, H. Bundgaard, 8, 1-38, (1992); J. Phann. Sci., 77, 285 (1988); Chem. Pharm. Bull., N. Nakeya et al., 32, 692 (1984); Pro-drugs as Novel Delivery Systems, T. Higuchi and V. Stella, 14 A.C.S. Symposium Series, and Bioreversible Carriers in Drag Design, E.B. Roche, ed., American Pharmaceutical Association and Pergamon Press, 1987; J. Med. Chem., Vol. 47, No. 10, 1-12 (2004), які шляхом посилання на них включаються у цей документ. Прикладом проліків сполуки винаходу є ефірні проліки. «Ефірні проліки» означають сполуку, яка може перетворюватися in vivo у ході метаболічних процесів (наприклад, за допомогою гідролізу) у сполуку винаходу. Наприклад, ефірні проліки сполуки винаходу, що містять карбоксигрупу, можуть перетворюватися за допомогою гідролізу in vivo у відповідну сполуку винаходу, наприклад, проліки на основі метилового ефіру, проліки на основі етилового ефіру або проліки на основі 2диметиламіноетилового ефіру. «Фармацевтично прийнятні солі» означає нетоксичні, неорганічні і органічні солі сполук, що складають предмет даного винаходу, утворені при додаванні кислоти або основи. Ці солі можуть бути одержані in situ на кінцевій стадії виділення і очищення сполук. «Фармацевтично ефективна кількість» означає кількість сполуки або сполук, які відповідно до даного винаходу ефективні для створення бажаного терапевтичного ефекту, описаного у цьому документі, наприклад зняття алергічної реакції або запалення. 95303 10 «Сольват» означає сполуку, що складає предмет даного винаходу, фізично зв'язану з однією або декількома молекулами розчинника. До фізичного зв'язування належить утворення водневого зв'язку. У визначених випадках сольват можна буде виділити, наприклад, коли одна або більше молекул розчинника включені у кристалічну решітку твердої кристалічної речовини. Термін «сольват» поширюється як на фазу розчину, так і на сольвати, що виділяються. Типовими сольватами є гідрати, етаноляти і метаноляти. Деякі зі сполук, що складають предмет даного винаходу, є основними, і такі сполуки застосовні у формі вільних основ або у формі їх фармацевтично прийнятних солей, утворених при додаванні кислоти. Солі, утворені при додаванні кислоти, можуть бути більш зручною формою для використання, і на практиці використання сольової форми по суті означає застосування форми вільної основи. До кислот, які можуть використовуватися для приготування солей, належать переважно такі, які при змішуванні з вільною основою приводять до фармацевтично прийнятних солей, тобто солей, аніони яких є нетоксичними для пацієнта у фармакологічних дозах солей, так що корисні ефекти інгібування, властиві вільній основі, не спотворюються побічними ефектами, що приписуються аніонам. Хоча фармацевтично прийнятні солі вказаних основних сполук є переважними, всі солі, утворені при додаванні кислот, являють собою корисні джерела форми вільної основи, навіть якщо конкретна сіль, сама по собі, потрібна тільки лише як проміжний продукт, наприклад, якщо сіль одержують виключно з метою очищення та ідентифікації, або якщо вона використовується як проміжна сполука при одержанні фармацевтично прийнятної солі за допомогою іонообмінних процесів. Зокрема, солі, утворені при додаванні кислот, можуть бути одержані незалежною реакцією очищеної сполуки у формі вільної основи з придатною органічною або неорганічною кислотою і виділенням одержаної таким чином солі. Фармацевтично прийнятні солі, які стосуються даного винаходу, включають солі, одержані з мінеральних і органічних кислот. Приклади солей, утворених при додаванні кислот, включають гідроброміди, гідрохлориди, сульфати, бісульфати, фосфати, нітрат, ацетати, оксалати, валерати, олеати, пальмітати, хінати, стеарати, лаурати, борати, бензоати, лактати, фосфати, тозилати, цитрати, малеати, фумарати, сукцинати, тартрати, нафтилати, мезилати, глюкогептонати, лактіобіонати, сульфамати, малонати, саліцилати, пропіонати, метилен-біс-гідроксинафтоати, гентисати, ізетіонати, ди-паратолуоїлтартрати, етансульфонати, бензолсульфонати, циклогексилсульфамати і лаурилсульфонати. Див., наприклад, роботу S.M. Berge, et al., "Pharmaceutical Salts, " J. Pharm. Sci., 66, 1-19 (1977), яка включена у даний документ як посилання. Якщо сполука, що складає предмет даного винаходу, заміщена кислотною групою, можуть одержуватися солі, що утворюються при додаванні основи, які є просто більш зручною формою засто 11 сування, і на практиці використання сольової форми по суті рівносильне її використанню у формі вільної кислоти. До основ, які можуть використовуватися для приготування солей, належать переважно такі, які при змішуванні з вільною кислотою приводять до фармацевтично прийнятних солей, тобто солей, катіони яких є нетоксичними для пацієнта у фармакологічних дозах солей, так що корисні ефекти інгібування, властиві вільній основі, не спотворюються побічними ефектами, що приписуються катіонам. Солі, утворені при додаванні основи, можна також одержати незалежним чином внаслідок реакції очищеної сполуки в її кислій формі з придатною органічною або неорганічною основою, що утворюється з солей лужних або лужноземельних металів, і виділенням одержаної таким чином солі. До солей, що утворюються при додаванні основи, належать фармацевтично прийнятні солі металів і амінів. До придатних солей металів належать солі натрію, калію, кальцію, барію, цинку, магнію і алюмінію. У конкретному прикладі солями є натрієві і калієві солі. Придатні неорганічні солі, що утворюються при додаванні основи, одержують з основ металів, до яких належать гідрид натрію, гідроксид натрію, гідроксид калію, гідроксид кальцію, гідроксид алюмінію, гідроксид літію, гідроксид магнію, гідроксид цинку і їм подібні. Придатні амінні солі, що утворюються при додаванні основи, одержують з амінів, основність яких достатня для утворення стабільної солі, і, переважно, з амінів, які часто використовуються у медичній хімії внаслідок їх низької токсичності і придатності для використання у медичних цілях, наприклад аміак, етилендіамін, N-метилглюкамін, лізин, аргінін, орнітин, холін, Ν,Ν'дибензилетилендіамін, хлорпрокаїн, діетаноламін, прокаїн, N-бензилфенетиламін, діетиламін, піперазин, трис(гідроксиметил)амінометан, гідроксид тетраметиламонію, триетиламін, дибензиламін, ефенамін, дегідроабіетиламін, N-етилпіперидин, бензиламін, тетраметиламоній, тетраетиламоній, метиламін, диметиламін, триметиламін, етиламін, основні амінокислоти, наприклад лізин і аргінін, і дициклогексиламін. Являючись корисними самі по собі як активні сполуки, солі сполук, що складають предмет даного винаходу, корисні для цілей очищення сполук, наприклад за рахунок використання різниці у розчинності солей і вихідних сполук, побічних продуктів і/або вихідних матеріалів за допомогою відомих фахівцям методів. Очевидно, що сполуки, які складають предмет даного винаходу, можуть містити асиметричні центри. Ці асиметричні центри можуть незалежно один від одного бути в R- або в S-конфігурації. Для фахівця у галузі техніки очевидно, що визначені сполуки, які складають предмет даного винаходу, можуть також проявляти геометричну ізомерію. Потрібно розуміти, що даний винахід поширюється на окремі геометричні ізомери і стереоізомери і їх суміші, у тому числі рацемічні суміші сполук винаходу. Такі ізомери можна виділити з їх сумішей за допомогою відомих методів або їх модифікацій, наприклад хроматографічних методів або методів перекристалізації, або одержати окремо з відпові 95303 12 дних ізомерів проміжних сполук. Крім того, у ситуаціях, де можливі таутомери сполук винаходу, даний винахід включає всі таутомерні форми таких сполук. Сполуки, що складають предмет даного винаходу, а також проміжні і вихідні речовини, що використовуються для їх одержання, називають відповідно до номенклатури ІЮПАК, в якій характеристичні групи при найменуванні мають наступний пріоритет за зменшенням важливості: кислоти, ефіри, аміди та ін. Проте, вважається, що якщо для якої-небудь зі сполук, представленої як структурною формулою, так і номенклатурною назвою, є невідповідність між структурною формулою і номенклатурною назвою, правильною необхідно вважати структурну формулу. Одним конкретним здійсненням даного винаходу є сполука формули (І), дe 1 2 R являє собою R CH2SO2-, R CH2SO2NH- або 3 R NHSO2-; 1 R являє собою феніл, необов'язково заміщений галогеном; 2 R являє собою феніл, заміщений галогеном; і 3 R являє собою 2,6-дихлорбензил, 3,5дихлорбензил, 2,4-дихлорфенілетил, 2метоксифенілетил, 3-метоксифенілетил, 4метоксифенілетил, 2-трифторметил-фенілетил, фенілетил або 3-феніл-н-пропіл, або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків. Ще одним конкретним здійсненням даного винаходу є сполука формули (І), де R являє собою 3 R NHSO2-, її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків. Ще одним конкретним здійсненням даного винаходу є сполука формули (II), де 4 R являє собою 4-хлор, 4-фтор, 4-метил або 7хлор; 5 R являє собою хлор або етил і 6 R являє собою водень або метил; 13 або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків. Ще одним конкретним здійсненням даного ви5 находу є сполука формули (II), де R являє собою 6 хлор і R являє собою водень, або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків. Ще одним конкретним здійсненням даного винаходу є сполука формули (III), 7 де R являє собою метил, метилсульфоніл, етилсульфоніл, галогеналкіл-сульфоніл або тетразоліл, або її фармацевтично прийнятна сіль, гідрат або сольват, її фармацевтично прийнятні проліки або фармацевтично прийнятна сіль, гідрат або сольват проліків. Ще одним конкретним здійсненням даного винаходу є сполука, що вибирається з наведеного нижче: {2-[4-хлор-3-(2,6дихлорбензилсульфамоїл)феніл]-1Н-індол-3іл}оцтової кислоти, {2-[4-хлор-3-(3,5дихлорбензилсульфамоїл)феніл]-1Н-індол-3іл}оцтової кислоти, (2-{4-хлор-3-[2-(2,4дихлорфеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти, (2-{4-хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти, (2-{4-хлор-3-[2-(3метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти, (2-{4-хлор-3-[2-(4метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти, (2-{4-хлор-3-[2-(2трифторметоксифеніл)етилсульфамоїл]феніл}-1Ніндол-3-іл)оцтової кислоти, [2-(4-хлор-3-фенетилсульфамоїлфеніл)-1Ніндол-3-іл]оцтової кислоти, {2-[4-хлор-3-(3-фенілпропілсульфамоїл)феніл]1Н-індол-3-іл}оцтової кислоти, 2-[2-(4-хлор-3-циклогексилсульфамоїлфеніл)1Н-індол-3-іл]-N-метилацетаміду, [4-хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти, калій[4-хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]ацетату, 95303 14 [2-(4-хлор-3-циклогексилсульфамоїлфеніл)-4фтор-1Н-індол-3-іл]оцтової кислоти, [2-(4-хлор-3-циклогексилсульфамоїлфеніл)-4метил-1Н-індол-3-іл]оцтової кислоти, [7-хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти, 2-хлор-N-циклогексил-5-[3-(2метансульфоніламіно-2-оксоетил)-1Н-індол-2іл]бензолсульфаміду, 2-хлор-N-циклогексил-5-[3-(2етансульфоніламіно-2-оксоетил)-1Н-індол-2іл]бензолсульфаміду, 2-хлор-N-циклогексил-5-[3-(2-оксо-2трифторметансульфоніламіноетил)-1Н-індол-2іл]бензолсульфаміду, 2-[2-(4-хлор-3-циклогексилсульфамоїлфеніл)1Н-індол-3-іл]-N-(1Н-тетразол-5-іл)ацетаміду, [2-(3-циклогексилсульфамоїл-4-етилфеніл)1Н-індол-3-іл]оцтової кислоти, 2-[2-(4-хлор-3-циклогексилсульфамоїлфеніл)1Н-індол-3-іл]пропіонової кислоти, {2-[4-хлор-3-(3хлорфенілметансульфоніл)феніл]-1Н-індол-3іл}оцтової кислоти або {2-[4-хлор-3-(3хлорфенілметансульфоніламіно)феніл]-1Н-індол3-іл}оцтової кислоти, або її фармацевтично прийнятної солі, гідрату або сольвату, її фармацевтично прийнятних проліків або фармацевтично прийнятної солі, гідрату або сольвату проліків. Сполуки, що складають предмет даного винаходу, проявляють активність як антагоністи рецептора простагландину D2 і можуть використовуватися як активні фармакологічні речовини. Відповідно, вони включаються у фармацевтичні композиції і використовуються у лікуванні пацієнтів, які страждають визначеними медичними порушеннями. Сполуки в обсязі даного винаходу є антагоністами рецептора простагландину D2 відповідно до тестів, описаних у літературі, а також описаних у наведеному нижче розділі про фармакологічні тести, результати яких, як вважається, корелюють з фармакологічною дією в організмі людини та інших ссавців. Таким чином, у ще одному здійсненні даного винаходу представлені сполуки, що складають предмет даного винаходу, і композиції, що містять їх, які можуть використовуватися у лікуванні пацієнтів, які страждають захворюваннями або схильні до захворювань, що можна полегшити введенням антагоніста PGD2. Наприклад, сполуки даного винаходу тому можуть використовуватися для лікування різних захворювань, опосередкованих PGD2, у тому числі, зокрема, алергічного захворювання (наприклад, алергічний риніт, алергічний кон'юнктивіт, атопічний дерматит, бронхіальна астма і харчова алергія), системного мастоцитозу, порушень, що супроводжуються системною активацією мастоцитів, анафілактичного шоку, бронхоконстрикції, бронхіту, кропивниці, екземи, захворювань, що супроводжуються свербежем (наприклад, атопічний дерматит і кропивниця), захворювань (наприклад, катаракта, відшарову 15 вання сітківки, запалення, інфекція і порушення сну), які виникають як вторинні захворювання внаслідок поведінки, що супроводжується свербежем (наприклад, розчісування і розтирання), запалення, хронічних обструктивних захворювань легенів, ішемічного реперфузійного ушкодження, розладу мозкового кровообігу, хронічного ревматоїдного артриту, плевриту, неспецифічного виразкового коліту і подібних захворювань. Крім того, сполуки, що складають предмет даного винаходу, можуть використовуватися для лікування у поєднанні з: (і) антигістамінами, наприклад, фексофенадин, лоратадин і цитиризин, для лікування алергічного риніту; (іі) антагоністами лейкотриєну, наприклад, монтелукаст і зафірлукаст, для лікування алергічного риніту, ХОЗЛ, алергічного дерматиту, алергічного кон'юнктивіту та ін. - точну інформацію див. у WO 01/78697 А2; (ііі) бета-агоністами, наприклад, альбутерол, сальбутерол і тербуталін, для лікування астми, ХОЗЛ, алергічного дерматиту, алергічного кон'юнктивіту та ін.; (iv) антигістамінами, наприклад, фексофенадин, лоратадин, деслоратадин і цетиризин, для лікування астми, ХОЗЛ, алергічного дерматиту, алергічного кон'юнктивіту та ін.; (ν) інгібіторами PDE4 (фосфодіестерази 4), наприклад, рофлуміласт і циломіласт, для лікування астми, ХОЗЛ, алергічного дерматиту, алергічного кон'юнктивіт та ін.; або (vi) з антагоністами ТР (рецептора тромбоксану А2) або антагоністами CrTh2 (молекули, гомологічної рецептору хемоатрактанту, що експресується на Th2 клітинах), наприклад, раматробран (BAY-u3405), для лікування ХОЗЛ, алергічного дерматиту, алергічного кон'юнктивіту та ін. Особливим здійсненням терапевтичних способів, що складають предмет даного винаходу, є лікування алергічного риніту. Ще одним особливим здійсненням терапевтичних способів, що складають предмет даного винаходу, є лікування бронхіальної астми. Відповідно до ще одного пункту винаходу пропонується спосіб лікування пацієнта (людини або тварини), який страждає захворюваннями, або схильний до захворювань, які можуть бути полегшені введенням антагоніста рецептора простагландину D2, наприклад захворюваннями, описаними вище, що включає введення пацієнту ефективної кількості сполуки, що складає предмет даного винаходу, або складу, що містить її. Під «ефективною кількістю» мається на увазі кількість сполуки, що складає предмет даного винаходу, яка ефективна як антагоніст рецептора простагландину D2 і тому здатна надавати бажаний терапевтичний ефект. Включені в опис посилання на лікування поширюються як на профілактичну терапію, так і на лікування діагностованих захворювань. Даний винахід поширюється також на фармацевтичні композиції, які включають, щонайменше, одну сполуку, що складає предмет даного винаходу, у суміші з фармацевтично прийнятним носієм. 95303 16 На практиці сполуки, що складають предмет даного винаходу, можуть вводитися у вигляді фармацевтично прийнятних лікарських форм людині та іншим тваринам за допомогою місцевого або системного застосування, у тому числі перорального, інгаляційного, ректального, назального, букального, сублінгвального, вагінального, кишкового, парентерального (у тому числі підшкірного, внутрішньом'язового, внутрішньовенного, внутрішньошкірного, інтратекального і епідурального), інтрацистернального і внутрішньочеревинного. Потрібно приймати до уваги, що переважний спосіб введення може варіюватися, наприклад, в залежності від стану пацієнта. «Фармацевтично прийнятними лікарськими формами» називаються лікарські форми сполуки, що складає предмет даного винаходу, які включають, наприклад, таблетки, драже, порошки, еліксири, сиропи, рідкі склади, у тому числі суспензії, спреї, інгалянти, таблетки, коржики, емульсії, розчини, гранули, капсули і супозиторії, а також рідкі склади для ін'єкцій, у тому числі ліпосомні препарати. Загальний опис методів і складів можна знайти в останньому виданні Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, PA. Особливим аспектом даного винаходу є сполука, що складає предмет даного винаходу, яка повинна вводитися у формі лікарського препарату. Відповідно до даного винаходу, фармацевтичні композиції складаються зі сполук, що складають предмет даного винаходу, і фармацевтично прийнятних носіїв. Фармацевтично прийнятні носії мають у своєму складі принаймні один з компонентів, якими можуть бути фармацевтично прийнятні носії, розріджувачі, оболонки, ад'юванти, формоутворювальні наповнювачі або середовища, такі як консерванти, наповнювачі, розпушувачі, змочувальні речовини, емульгатори, стабілізатори емульсій, суспендуючі речовини, ізотонічні речовини, підсолоджувачі, смакові домішки, ароматизатори, барвники, бактерицидні засоби, протигрибкові засоби, інші терапевтичні речовини, змащувальні речовини, речовини, що сповільнюють або прискорюють всмоктування, і дозуючі речовини, в залежності від особливостей способу введення і лікарської форми. Прикладами суспендуючих речовин є етоксильовані ізостеарилові спирти, поліоксіетиленсорбіт і складні ефіри сорбітану, мікрокристалічна целюлоза, метагідроксид алюмінію, бентоніт, агар-агар і трагакант або суміші цих речовин. Прикладами бактерицидних і протигрибкових речовин, що запобігають дії мікроорганізмів, є парабени, хлорбутанол, фенол, сорбінова кислота і т.п. Прикладами ізотонічних речовин є цукор, хлорид натрію і т.п. Прикладами речовин, що сповільнюють і прискорюють всмоктування, є моностеарат алюмінію і желатин. Прикладами речовин, що прискорюють і стимулюють абсорбцію, є диметилсульфоксид і його аналоги. Прикладами розріджувачів, розчинників, носіїв, солюбілізуючих домішок, емульгаторів і стабілі 17 заторів емульсії є вода, хлороформ, сахароза, етанол, ізопропіловий спирт, етиловий ефір вугільної кислоти, етилацетат, бензиловий спирт, тетрагідрофурфуриловий спирт, бензилбензоат, поліоли, пропіленгліколь, 1,3-бутиленгліколь, гліцерин, поліетиленгліколі, диметилформамід, Tween 60, Span 60, цетостеариловий спирт, міристиловий спирт, гліцерилмоностеарат і лаурилсульфат натрію, складні ефіри сорбітану і жирних кислот, рослинні олії (такі як бавовняна олія, арахісова олія, олія пшеничних зародків, оливкова олія, касторова олія і кунжутна олія) і органічні складні ефіри, що ін'єктуються, такі як етилолеат і йому подібні, або придатні суміші цих сполук. Прикладами формоутворювальних наповнювачів є лактоза, молочний цукор, цитрат натрію, карбонат кальцію і дикальційфосфат. Прикладами розпушувачів є крохмаль, альгінові кислоти і деякі складні силікати. Прикладами ковзних речовин є стеарат магнію, лаурилсульфат натрію, тальк, а також поліетиленгліколі з високою молекулярною вагою. Вибір фармацевтично прийнятного носія, загалом, визначається відповідно до хімічних властивостей активної сполуки, таких як розчинність, метод введення і норми, яких необхідно дотримуватися у фармацевтичній практиці. Фармацевтичні композиції, що складають предмет даного винаходу, придатні для перорального застосування, можуть являти собою лікарські засоби, такі як тверді лікарські форми, такі як капсули, облатки або таблетки, кожна з яких містить визначену кількість активного інгредієнта, або такі як порошки або гранули, або рідкі лікарські форми, такі як розчини або суспензії у водному або неводному рідкому середовищі, або рідкій емульсії масло-у-воді або вода-у-маслі. Активний інгредієнт також може бути у вигляді болюсу, електуарію або пасти. «Тверда лікарська форма» означає лікарську форму сполуки, що складає предмет даного винаходу, у вигляді твердої речовини, наприклад капсули, таблетки, пілюлі, порошку, драже або гранули. У таких лікарських формах сполука, що складає предмет даного винаходу, додана в як мінімум один традиційно використовуваний інертний формоутворювальний наповнювач (або носій), наприклад, цитрат натрію або дикальційфосфат, або (а) наповнювачі або середовища, такі як, наприклад, крохмаль, лактоза, сахароза, глюкоза, маніт і кремнієва кислота, (b) зв'язувальні речовини, наприклад карбоксиметилцелюлоза, альгінати, желатин, полівінілпіролідон, сахароза і камедь, (с) зволожуючі речовини, такі як, наприклад, гліцерин, (d) розпушувачі, такі як, наприклад, агар-агар, карбонат кальцію, картопляний або тапіоковий крохмаль, альгінова кислота, визначені складні силікати і Na2CO3, (e) уповільнювачі утворення розчинів, такі як, наприклад, парафін, (f) прискорювачі абсорбції, такі як, наприклад, четвертинні амонієві сполуки, (g) зволожуючі речовини, такі як, наприклад, цетиловий спирт і гліцеринмоностеарат, (h) адсорбенти, такі як, наприклад, каолін або бентоніт, (і) ковзні речовини, такі як, наприклад, тальк, стеарат кальцію, стеарат магнію, тверді поліетиленгліколі, 95303 18 лаурилсульфат натрію, (j) скаламучувальні компоненти, (k) буферні речовини і речовини, що вивільняють сполуку або сполуки, що складають предмет даного винаходу, з відстрочкою у визначеній частині кишкового тракту. Таблетка може бути приготована пресуванням або формуванням і може мати один або декілька допоміжних компонентів. Пресовані таблетки можна одержувати пресуванням у придатному апараті активного інгредієнта у сипкій формі, такій як порошок або гранули, необов'язково змішаній зі зв'язувальною речовиною, ковзною речовиною, інертним розріджувачем, консервантом, поверхневоактивною або диспергуючою речовиною. Можуть використовуватися такі інертні наповнювачі як лактоза, цитрат натрію, карбонат кальцію, дикальційфосфат і розпушувачі, такі як крохмаль, альгінові кислоти і визначені складні силікати, змішані з ковзними речовинами, такими як стеарат магнію, лаурилсульфат натрію і тальк. Суміш порошкоподібних сполук, змочену інертним рідким розріджувачем, можна формувати на придатному апараті для одержання формованих таблеток. Таблетки можуть мати покриття або насічки, а також можуть мати склад, що забезпечує повільне або контрольоване вивільнення активного інгредієнта, що міститься у них. Тверді склади також можуть використовуватися як наповнювачі у желатинових капсулах з м'яким або твердим наповненням з використанням таких інертних наповнювачів як лактоза або молочний цукор, а також поліетиленгліколів з великою молекулярною вагою і подібних їм речовин. У випадку необхідності, а також для більш ефективного поширення, сполуки можуть бути мікроінкапсульовані у системах повільного або направленого вивільнення, таких як біосумісні, біорозкладані полімерні матриці (наприклад, співполімер с1,1-лактиду з гліколідом), ліпосоми і мікросфери, або приєднані до них, а потім підшкірно і внутрішньом'язово ін'єктовані методом, називаним підшкірною або внутрішньом'язовою ін'єкцією уповільненого всмоктування, що забезпечує повільне безперервне вивільнення сполуки або сполук протягом 2 тижнів або довше. Сполуки можуть бути стерилізовані, наприклад фільтрацією через фільтр, що затримує бактерії, або додаванням стерилізуючих речовин у формі стерильних твердих складів, які можуть бути розчинені у стерильній воді або іншому стерильному середовищі, що ін'єктується, безпосередньо перед застосуванням. «Рідка лікарська форма» означає форму активної сполуки, яка вводиться пацієнту у рідкій формі, наприклад, у вигляді фармацевтично прийнятних емульсій, розчинів, суспензій, сиропів і еліксирів. Крім активних сполук, рідкі лікарські форми можуть містити звичайно застосовувані у даній галузі інертні розріджувачі, такі як розчинники, солюбілізуючі речовини і емульгатори. При використанні водних суспензій вони можуть містити емульгатори або речовини, які сприяють утворенню суспензій. Фармацевтичні композиції, придатні для місцевого застосування, представляють склади у формі, що допускає місцеве введення пацієнту. 19 Склади можуть мати форму мазей місцевого застосування, бальзамів, порошків, спреїв та інгалянтів, гелів (на водній або спиртовій основі), кремів, що звичайно використовуються у даній галузі, або бути включеними у матричну основу для застосування як пластир, що забезпечує контрольоване вивільнення сполуки через шкірний бар'єр. У формі мазі активні інгредієнти можуть використовуватися з парафіновими або водорозчинними основами. В альтернативному варіанті активні інгредієнти можуть мати форму крему з масляноводною основою. Склади, призначені для місцевого застосування в око, являють собою очні краплі, в яких активний інгредієнт розчинений або зважений у придатному носії, як правило, водному розчиннику. Склади, призначені для місцевого застосування через слизову рота, включають лікарські льодяники, що мають у своєму складі активний інгредієнт у смаковій основі, як правило, сахарозі і камеді або трагаканті; пастилки, що мають у своєму складі активний інгредієнт в інертній основі, такий як желатин і гліцерин або сахароза і камедь; і склади для полоскання рота, що мають у своєму складі активний інгредієнт у придатному рідкому носії. Масляна фаза емульсійних фармацевтичних композицій може бути одержана звичайним способом з відомих інгредієнтів. Ця фаза може мати у своєму складі тільки емульгатор (також називаний емульгуючою речовиною), однак бажано, щоб до неї також входив як мінімум один емульгатор, що містить жир або масло, або емульгатор, що містить і жир, і масло. В одному конкретному здійсненні винаходу у склад включений гідрофільний емульгатор поряд з ліпофільним емульгатором, який діє як стабілізатор. Емульгатор(и) разом зі стабілізатором(ами) або без нього утворюють віск, що емульгується, а разом з маслом або жиром утворюють основу мазі, що емульгується, яка є масляною диспергованою фазою кремових складів. За бажанням водна фаза кремової основи може включати, наприклад, не менше 30% ваг./ваг. багатоатомного спирту, тобто спирту з двома або більше гідроксильними групами, наприклад пропіленгліколь, бутан-1,3-діол, манітол, сорбітол, гліцерин і поліетиленгліколь (у тому числі PEG 400) і їх суміші. Склади для місцевого застосування можуть, якщо необхідно, мати у своєму складі сполуку, що стимулює всмоктування або проникнення активного інгредієнта через шкіру або інші уражені зони. Вибір придатних масел або жирів для використання у складі залежить від властивостей, які необхідно одержати. Бажано, щоб крем був нежирним продуктом придатної консистенції, що не залишає плям і змивається, і не допускає витоку з тюбиків та інших ємностей. Можуть використовуватися лінійні і розгалужені одно-і двохосновні алкілові ефіри, такі як діізопропілміристат, децилолеат, ізопропілпальмітат, бутилстеарат, 2етилгексилпальмітат або суміш складних ефірів з розгалуженим ланцюгом, відома як Crodamol CAP. Вони можуть використовуватися окремо або у сумішах, в залежності від бажаних властивостей. Як 95303 20 альтернатива можуть використовуватися ліпіди з високою температурою плавлення, такі як білий м'який парафін і/або рідкий парафін та інші мінеральні масла. Фармацевтичними композиціями для ректального або вагінального застосування називаються склади, форма яких допускає ректальне або вагінальне введення пацієнту, і які містять як мінімум одну сполуку, що складає предмет даного винаходу. Супозиторії являють собою одну з форм таких складів, яку можна одержати змішуванням сполук, що складають предмет даного винаходу, з придатними неподразнюючими інертними середовищами або носіями, наприклад, олія какао, поліетиленгліколь або воскова основа супозиторію, які знаходяться у твердому стані при звичайних температурах, але стають рідкими при температурі тіла і тому плавляться при ректальному або вагінальному введенні і вивільняють активний інгредієнт. Фармацевтичні композиції, що вводяться за допомогою ін'єкції, можуть вводитися внутрішньом'язово, внутрішньовенно, внутрішньочеревинно і/або підшкірно. Склади, що складають предмет даного винаходу, готують у рідких розчинах, зокрема у фізіологічно сумісних буферах, таких як розчин Хенка або розчин Рінгера. Крім того, склади можуть бути приготовані у твердій формі і розчинені або зважені безпосередньо перед застосуванням. Можливі також ліофілізовані форми. Склади є стерильними і включають емульсії, суспензії, водні і неводні розчини для ін'єкцій, які можуть містити суспендуючі речовини, загусники і антиоксиданти, буфери, бактеріостати і домішки, що роблять склад ізотонічним, і його рН повинен бути приведений у відповідність з кров'ю пацієнта, якому буде вводитися препарат. Фармацевтичними композиціями, що складають предмет даного винаходу, придатними для назального або інгаляційного застосування, називаються склади, форма яких придатна для введення пацієнту назально або інгаляційно. Склад може містити носій у формі порошку з розмірами частинок, наприклад, у діапазоні від 1 до 500 мікрон (у тому числі з розмірами частинок у діапазоні від 20 до 500 мікрон з кроком 5 мікрон, тобто з розмірами 30 мікрон, 35 мікрон і т.д.). До придатних складів з рідким носієм для застосування, наприклад, як спрею або крапель для носа, належать водні або масляні розчини активного інгредієнта. Склади, придатні для аерозольного введення, можуть бути одержані відповідно до традиційних методів і можуть вводитися з іншими терапевтичними речовинами. Для введення складів, що складають предмет даного винаходу, як інгаляційна терапія можуть використовуватися дозуючі інгалятори. Дійсне дозування активного інгредієнта (інгредієнтів) у складах, що складають предмет даного винаходу, можна варіювати з метою одержання кількості активного інгредієнта (інгредієнтів), ефективної для одержання бажаного терапевтичного ефекту для визначеного складу і методу його введення пацієнту. Тому дозування, що вибирається для кожного пацієнта, залежить від множини фак 21 торів, таких як бажаний терапевтичний ефект, спосіб введення, бажана тривалість лікування, етіологія і тяжкість захворювання, стан пацієнта, вага, стать, дієта і вік, тип і активність кожного активного інгредієнта, швидкість абсорбції, метаболізм і/або виділення та інших факторів. Повна денна доза сполук, які складають предмет даного винаходу, що вводиться пацієнту у вигляді однієї або декількох доз, може складати, наприклад, приблизно від 0,001 до 100 мг/кг маси тіла на день, переважно від 0,01 до 10 мг/кг/день. Наприклад, для дорослого дози, як правило, складають приблизно від 0,01 до 100, переважно приблизно від 0,01 до 10 мг/кг маси тіла на день при інгаляції, приблизно від 0,01 до 100, переважно від 0,1 до 70, бажано від 0,5 до 10 мг/кг маси тіла на день при пероральному введенні, і приблизно від 0,01 до 50, переважно від 0,01 до 10 мг/кг маси тіла на день при внутрішньовенному введенні. Процентний вміст активного інгредієнта може бути різним, але він повинен забезпечувати одержання придатного дозування. Лікарські форми можуть містити окремі одиниці дози, що дозволяють одержати бажану денну дозу. Природно, що форми, які містять декілька одиниць дози, можуть вводитися приблизно одночасно. Введення доз може бути настільки частим, наскільки необхідно для досягнення бажаного терапевтичного ефекту. Деякі пацієнти можуть проявляти швидку реакцію у відповідь на більш високу або більш низьку дозу, і для них можуть виявитися достатніми набагато більш слабкі підтримуючі дози. Для інших пацієнтів можуть знадобитися довгострокові курси лікування з частотою від 1 до 4 доз на день відповідно до фізіологічних потреб кожного конкретного пацієнта. Зрозуміло, що для інших пацієнтів може бути потрібно не більше однієї або двох доз на день. Стандартні дози складів можуть бути одержані будь-яким з традиційно використовуваних у фармації методів. Такі методи включають стадію зв'язування активного інгредієнта з носієм, що складається з одного або декількох допоміжних інгредієнтів. Як правило, склади одержують однорідним і ретельним зв'язуванням активного інгредієнта з рідкими носіями або дрібнозернистими твердими носіями, або і тими і іншими, з подальшим формуванням продукту, якщо це необхідно. Склади можуть бути в упаковках по одній дозі або по декілька доз, наприклад, запаяні ампули і пляшечки з еластичними пробками, і можуть зберігатися у ліофілізованому стані, що потребує тільки додавання стерильного рідкого носія, наприклад, води для ін'єкцій, безпосередньо перед застосуванням. Індивідуальні розчини для ін'єкцій і суспензії можуть бути приготовані зі стерильних порошків, гранул або таблеток описаних вище типів. Сполуки, що складають предмет даного винаходу, можуть бути одержані за допомогою застосування або адаптації відомих методів, під якими маються на увазі методи, що використовувалися раніше або описані у літературі, наприклад, описані у роботі R.C. Larock in Comprehensive Organic Transformations, VCH publishers, 1989. Ефірні проліки сполук винаходу можуть бути одержані поєднанням сполук винаходу, що мають 95303 22 у своєму складі карбоксигрупу, зі спиртом з формулою YOH (де Υ являє собою алкіл або алкіл, заміщений аміно, алкіламіно або діалкіламіно), щоб одержати ефірний зв'язок за допомогою стандартних методів поєднання. Приклади включають поєднання у присутності HBTU і, як варіант, у присутності DIEA, у DCM при кімнатній температурі. Відповідно до ще однієї ознаки винаходу, утворені при додаванні кислоти солі сполук, що складають предмет даного винаходу, можна одержати внаслідок реакції вільної основи з відповідною кислотою за допомогою відомих методів або їх модифікацій. Наприклад, утворені при додаванні кислоти солі сполук, що складають предмет даного винаходу, можна одержати або розчиненням вільної основи у воді або водному розчині спирту або інших придатних розчинниках, що містять відповідну кислоту, і виділенням солі випарюванням розчину, або внаслідок реакції вільної основи і кислоти в органічному розчиннику, де сіль випадає в осад або випарюється з розчину. Сполуки, що складають предмет даного винаходу, можна регенерувати з їх солей, утворених приєднанням кислоти за допомогою відомих методів або їх модифікацій. Наприклад, вихідні сполуки, що складають предмет даного винаходу, можна регенерувати з їх солей, утворених приєднанням кислоти, лугами, наприклад водним розчином бікарбонату натрію або водним розчином аміаку. Сполуки, що складають предмет даного винаходу, можна регенерувати з їх солей, утворених приєднанням основи, за допомогою відомих методів або їх модифікацій. Наприклад, вихідні сполуки, що складають предмет даного винаходу, можна регенерувати з їх солей, утворених приєднанням основи, обробкою кислотою, наприклад соляною кислотою. Сполуки, що складають предмет даного винаходу, можна без великих зусиль одержувати або утворювати у процесі, що складає предмет даного винаходу, як сольвати (наприклад, гідрати). Гідрати сполук, що складають предмет даного винаходу, можуть бути без великих зусиль одержані шляхом перекристалізації з суміші з водного/органічного розчинника за допомогою таких органічних розчинників як діоксан, THF або МеОН. Ще однією відмітною ознакою даного винаходу є одержання утворених при додаванні основи солей сполук, що складають предмет даного винаходу, внаслідок реакції вільної кислоти з відповідною основою за допомогою відомих методів або їх модифікацій. Наприклад, утворені при додаванні основи солі сполук, що складають предмет даного винаходу, можна одержати або розчиненням вільної кислоти у воді або водному розчині спирту або інших придатних розчинниках, що містять відповідну основу, і виділенням солі випарюванням розчину, або внаслідок реакції вільної кислоти і основи в органічному розчиннику, де сіль випадає в осад або випарюється з розчину. Вихідні або проміжні сполуки можна одержати за допомогою способів, описаних у даній заявці або адаптацією відомих способів. 23 Сполуки, що складають предмет даного винаходу, способи їх одержання, а також: їх біологічна активність будуть більш очевидні з розгляду наведених далі прикладів, які наводяться тільки як ілюстрація і не повинні розглядатися як такі, що обмежують обсяг винаходу. Сполуки, що складають предмет даного винаходу, ідентифікували, наприклад, за допомогою наведених нижче аналітичних методів: рідинну хроматографію високого тиску і масспектрометрію (РХМС) для визначення часу утримування (RT) і відповідних мас іонів проводять з використанням одного з наведених далі способів: мас-спектри (МС) записують на масспектрометрі Micromass LCT. Метод включає іонізацію позитивним електророзпиленням і сканування маси m/z від 100 до 1000. Рідинну хроматографію здійснюють за допомогою бінарного насоса і дегазатора Hewlett Packard 1100 Series; нерухома фаза: колонка Phenomenex Synergi 2 Hydro-ΡνΡ 20x4,0 мм, рухома фаза: А=0,1% мурашина кислота (FA) у воді, В=0,1% FA в ацетонітрилі. Об'єм введення 5 мкл за допомогою системи СТС Analytical PAL. Швидкість потоку елюенту становить 1 мл/хв. Градієнт складає від 10% В до 90% В за 3 хвилини і від 90% В до 100% В за 2 хвилини. Допоміжні детектори: УФ детектор Hewlett Packard 1100 Series, довжина хвилі = 220 нм, і випарний детектор світлорозсіювання (ELS) Sedere SEDEX 75, температура детектора = 46°С, тиск N2=4 бар. Спектри ядерного магнітного резонансу (ЯМР) 1 300 МГЦ Н записують при кімнатній температурі на спектрометрі Varian Mercury (300 МГц) з 5-мм датчиком ASW. В ЯМР величини хімічного зсуву () вказані у мільйонних частках (м.ч.) відносно тетраметилсилану (TMS) як внутрішнього стандарту. У наведених далі прикладах і описах синтезу, а також в іншій частині заявки, використовувані терміни мають наведені нижче значення: «кг» кілограми, «г» - грами, «мг» - міліграми, «мкг, цг» мікрограми, «моль» - молі, «ммоль» - мілімолі, «М» - молярність, «мМ» - мілімолярність, «мкМ, Μ» - мікромолярність, «нМ» - наномолярність, «л» - літри, «мл» мілілітри, «мкл, л» - мікролітри, «°С» - градуси Цельсія, «т.пл.» - точка плавлення, «т.к.» - точка кипіння, «мм рт.ст.» - тиск у міліметрах ртутного стовпчика, «см» - сантиметри, «нм» нанометри, «абс.» - абсолютний, «конц.» - концентрований, «с» - концентрація у г/мл, «к.т.» - кімнатна температура, «ТШХ» - тонкошарова хроматографія, «ВЕРХ» - високоефективна рідинна хроматографія, «в/ч» - внутрішньочеревинно, «в/в» - внутрішньовенно, «с» - синглет, «д» - дублет; «т» - триплет; «кв» - квартет; «м» - мультиплет, «дд» - дублет дублетів; «уш.» - розширений, «РХ» - рідинний хроматограф, «МС» масспектрограф, «ESI/MS» - іонізація в електроспреї/мас-спектрограф, «RT» - час утримування, «М» - молекулярний іон, «PSI» - «фунтів на кв.дюйм» фунтів на квадратний дюйм, «DMSO» - диметилсульфоксид, «DMF» - Ν,Ν-диметилформамід, «CDI» - 1,1'-карбонілдіімідазол, «DCM» або «СН2Сl2» - дихлорметан, «HCl» - соляна кислота, «SPA» - SPA-аналіз (сцинтиляційний аналіз з ви 95303 24 користанням молекулярно імпринтованих полімерів), «АТТС» - Американська колекція типових культур, «FBS» - ембріональна бичача сироватка, «МПС» - мінімальне підтримуюче середовище, «імп./хв.» - кількість імпульсів за хвилину, «EtOAc» - етилацетат, «PBS» - фосфатний буферний фізіологічний розчин, «ТМД» - трансмембранний домен, «ІВМХ» - 3-ізобутил-1-метилксантин, «сАМР» - циклічний аденозинмонофосфат, «ІЮПАК» - Міжнародний союз теоретичної і прикладної хімії, «МГц» - мегагерц, «ПЕГ» - поліетиленгліколь, «МеОН» - метанол, «N» - нормальність, «THF» тетрагідрофуран, «год.» - година, «хв.» - хвилина(и), «MeNH2» - метиламін, «N2» - газоподібний азот, «iРrОН» -ізопропіловий спирт, «O.D.» - зовнішній діаметр, «MeCN» або «CH3CN» - ацетонітрил, «Et2O» - етиловий ефір, «TFA» - ТФК, «преп РХ» - препаративна рідинна флеш-хроматографія, «ТФЕ» - твердофазова екстракція, «LAH» - літійалюмінійгідрид, «пмол» - пікомолярність, «гептан» н-гептан, смола «НМВА-АМ» - амінометильна смола 4-гідроксиметилбензойної кислоти, «PdCl2(dppf)2» -комплекс дихлориду 1,1'біс(дифенілфосфіно)фероценпаладію(II) у DCM, «HBTU» 2-(1Н-бензотриазол-1-іл)-1,1,3,3тетраметилуроній гексафторфосфат, «DIEA» діізопропілетиламін, «CsF» - цезію фторид, «LiOH» - гідроксид літію, «~» - приблизно, «ІС50»» - концентрації сполуки, що дають 50% інгібування у SPAаналізі цАМФ у Т-клітинах LSI74 людини. Приклади Приклад 1 (а) {2-[4-хлор-3-(2,4дихлорбензилсульфамоїл)феніл]-1Н-індол-3іл}оцтова кислота Стадія 1. Димлячу азотну кислоту (1,5 л) охолоджують приблизно до -5°С у льодяній бані з сіллю. Протягом 30 хв. до перемішуваного механічною мішалкою розчину порціями додають 4-(4хлорфеніл)-4-оксомасляну кислоту (150 г, 0,706 моль) і реакційну суміш перемішують при температурі приблизно від -5°С до -7°С протягом 3,5 годин. Реакційну суміш виливають у воду з коленим льодом (3 л) і перемішують протягом ночі при кімнатній температурі. Твердий осад фільтрують, промивають водою до нейтральних змивів, сушать на повітрі, а потім досушують у вакуумній печі приблизно при 85°С для одержання 4-(4-хлор-3нітрофеніл)-4-оксомасляної кислоти у вигляді твердої речовини (159,1 г). Стадія 2. До перемішуваної механічною мішалкою суспензії 4-(4-хлор-3-нітрофеніл)-4оксомасляної кислоти (150 г, 0,582 моль) у воді (900 мл) і концентрованій HCl (12 мл) підливають розчин бісульфіту натрію (393 г, 2,07 моль, у 800 мл води) протягом 40 хвилин при 100-105°С. Після 25 додавання суміш нагрівають зі зворотним холодильником протягом 1 години. рН коректується до ~2 додаванням 4н HCl (100 мл). Суміш нагрівають зі зворотним холодильником ще 30 хвилин, доводять до кімнатної температури і фільтрують для одержання 4-(3-аміно-4-хлорфеніл)-4-оксомасляної кислоти у вигляді твердої речовини (79,3 г). 1 РХ/МС: RT=2,39 хв., МС: 228 (М+Н); H ЯМР (300 МГц, ДМСО-D6) 2,51 (т, J=6 Гц, 2H), 3,11 (т, J=6 Гц, 2H), 5,58 (с, 2Н), 7,1 (дд, J=6,2 Гц, J=2 Гц, 1Н), 7,29 (д, J=8 Гц, 1Н), 7,36 (д, J=2 Гц, 1Н), 12,08 (уш.с, 1Н). Стадія 3. 4-(3-Аміно-4-хлорфеніл)-4оксомасляну кислоту (16,2 г, 71,16 ммоль) у DMF (20 мл) додають до суміші концентрованої HCl (35 мл) і льоду (150 г). Розчин нітриту натрію (5,25 г, 76,1 ммоль) у воді (18 мл) підливають піпеткою, зануривши її кінець у розчин, протягом 5 хвилин при температурі від -5°С до -10°С. Реакційну суміш нагрівають до 0°С і перемішують протягом 15 хвилин. Одержаний розчин повільно додають при кімнатній температурі до суміші дигідрату хлориду міді (5,58 г, 32,7 ммоль) у льодяній оцтовій кислоті (175 мл), який був насичений газоподібним діоксидом сірки. Одержаний розчин перемішують протягом 45 хвилин при кімнатній температурі, додають воду (500 мл) і розчин перемішують протягом 1 години. Посудину охолоджують до 10°С і осад фільтрують та промивають водою для одержання 4(4-хлор-3-хлорсульфонілфеніл)-4-оксомасляної кислоти у вигляді твердої речовини (12,94 г). 1 РХ/МС: RT=2,68 хв., МС: 310 (М+Н); H ЯМР (300 МГц, ДМСО-D6) м.ч. 2,56 (т, J=6 Гц, 2Н), 3,19 (т, J=6 Гц, 2Н), 7,51 (д, J=8 Гц, 1Н), 7,87 (дд, J=6 Гц, J=2 Гц, 1Н), 8,39 (д, J=2 Гц, 1Н), 12,66 (уш.с, 1Н). Стадія 4. 4-(4-Хлор-3-хлорсульфонілфеніл)-4оксомасляну кислоту (2 г, 6,43 ммоль) додають до перемішуваного розчину 2,4-дихлорбензиламіну (2,82 г, 16 ммоль) у суміші DCM:MeOH (1:1, 50 мл) при 0°С. Реакційну суміш нагрівають до кімнатної температури і перемішують протягом 20 годин. Реакційну суміш підкислюють 2н водної HCl (рН ~2) і двічі екстрагують дихлорметаном. Об'єднану органічну фазу промивають водою, сольовим розчином, сушать над сульфатом натрію і випарюють у вакуумі для одержання 4-[4-хлор-3-(2,4дихлорбензилсульфамоїл)феніл]-4-оксомасляної кислоти у вигляді напівтвердої речовини (2,1 г). РХ/МС: RT=2,38 хв., МС: 448 (М-Н). Стадія 5. До суміші 4-[4-хлор-3-(2,4дихлорбензилсульфамоїл)феніл]-4-оксомасляної кислоти (800 мг, 1,78 ммоль), моногідрату птолуолсульфонової кислоти (520 мг, 2,7 ммоль) і хлориду цинку (370 мг, 2,7 ммоль) у льодяній оцтовій кислоті (15 мл) у мікрохвильовій посудині додають феніл гідразин (300 мг, 2,78 ммоль). Закриту посудину нагрівають у мікрохвильовій печі при температурі 180°С протягом 40 хвилин. Реакційну суміш розбавляють EtOAc, переносять у конічну колбу і додають водну 2н HCl (-50 мл). Органічний шар відділяють і водний шар екстрагують EtOAc. Об'єднану органічну фазу промивають водою, сольовим розчином, сушать над сульфатом натрію і концентрують. Осад очищають препаративним розділенням ВЕРХ (рухома фаза: ацетоні 95303 26 трил-вода з 0,1% ТФК; градієнт 10-100% протягом 10 хвилин) для одержання {2-[4-хлор-3-(2,4дихлорбензилсульфамоїл)феніл]-1Н-індол-3іл}оцтової кислоти у вигляді твердої речовини (145 1 мг). РХ/МС: RT=2,78 хв., МС: 523 (М+Н). H ЯМР (300 МГц, ДМСО-D6) 3,74 (с, 2Н), 4,23 (д, J=6 Гц, 2Н), 7,06 (т, J=7 Гц, 1Н), 7,17 (т, J=7 Гц, 1Н), 7,37,48 (м, 4Н), 7,56 (д, J=8 Гц, 1Н), 7,75 (д, J=8,3 Гц, 1Н), 7,87 (д, J=8 Гц, 1Н), 8,22 (д, J=2 Гц, 1Н), 8,64 (т, J=6,9 Гц, 1Н), 11,52 (с, 1Н), 12,4 (уш.с, 1Н). IС50=4 нМ. (b) {2-[4-Хлор-3-(2,6дихлорбензилсульфамоїл)феніл]-1Н-індол-3іл}оцтова кислота Стадія 1. Методом, аналогічним описаному у прикладі 1(а), стадія 4, але використовуючи 2,6дихлорбензиламін (2,82 г) замість 2,4дихлорбензиламіну, одержують 4-[4-хлор-3-(2,6дихлорбензилсульфамоїл)феніл]-4-оксомасляну кислоту у вигляді порошку (2,12 г). РХ/МС: RT=2,1 хв., МС: 448 (М+Н). Стадія 2. Методом, аналогічним описаному на стадії 5 методу А у прикладі 1(а), але використовуючи 4-[4-хлор-3-(2,6дихлорбензилсульфамоїл)феніл]-4-оксомасляну кислоту (0,8 г) замість 4-[4-хлор-3-(2,4дихлорбензил-сульфамоїл)феніл]-4-оксомасляної кислоти, одержують {2-[4-хлор-3-(2,4дихлорбензилсульфамоїл)феніл]-1Н-індол-3іл}оцтову кислоту у вигляді твердої речовини (80 1 мг). РХ/МС: RT=2,72 хв, МС: 523 (М+Н); H ЯМР (300 МГц, ДМСО-D6) 3,75 (с, 2Н), 4,36 (д, 2Н, J=5,2 Гц), 7,06 (т, J=7 Гц, 1H), 7,1-7,45 (м, 5Н), 7,73 (д, J=8,5 Гц, 1Н), 7,57 (д, J=8 Гц, 1Н), 7,88 (дд, J=6 Гц, J=2,2 Гц, 1Н), 8,25 (д, J=2 Гц, 1Н), 8,33 (т, J=5 Гц, 1Н), 11,5 (с, 1Н), 12,4 (уш.с, 1Н). IС50=3 нМ. (c) {2-[4-Хлор-3-(3,5дихлорбензилсульфамоїл)феніл]-1Н-індол-3іл}оцтова кислота Стадія 1. Методом, аналогічним описаному на стадії 4 у прикладі 1(а), але використовуючи 3,5дихлорбензиламін (2,82 г) замість 2,4дихлорбензиламіну, одержують 4-[4-хлор-3-(3,5дихлорбензилсульфамоїл)феніл]-4-оксомасляну кислоту у вигляді твердої речовини (2,12 г). РХ/МС: RT=2,42 хв., МС: 450 (М+Н). Стадія 2. Методом, аналогічним описаному на стадії 5 методу А у прикладі 1(а), але використовуючи 4-[4-хлор-3-[3,5 27 дихлорбензилсульфамоїл)феніл]-4-оксомасляну кислоту (0,8 г) замість 4-[4-хлор-3-(2,4дихлорбензил-сульфамоїл)феніл]-4-оксомасляної кислоти, одержують {2-[4-хлор-3-(3,5дихлорбензилсульфамоїл)феніл]-1Н-індол-3іл}оцтову кислоту у вигляді твердої речовини (160 1 мг). РХ/МС: RT=2,78 хв., МС: 523 (М+Н). Н ЯМР (300 МГц, ДМСО-D6) 3,72 (с, 2Н), 4,19 (д, J=6,2 Гц, 2Н), 7,06 (т, J=7 Гц. 1Н), 7,1-7,45 (м, 5Н), 7,57 (д, J=8 Гц, 1Н), 7,71 (д, J=8,2 Гц, 1Н), 7,85 (дд, J=6,2 Гц, J=2,2 Гц, 1Н), 8,19 (д, J=2,2 Гц, 1Н), 8,65 (т, J=6,4 Гц, 1Н), 11,5 (с, 1Н), 12,4 (уш.с, 1Н). IС50=12 нМ. (d) (2-{4-Хлор-3-[2-(2,4дихлорфеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтова кислота Стадія 1. Методом, аналогічним описаному на стадії 4 у прикладі 1(а), але використовуючи 2,4дихлорфенетиламін (3,04 г) замість 2,4дихлорбензиламіну, одержують 4-{4-хлор-3-[2-(2,4дихлорфеніл)етилсульфамоїл]феніл}-4оксомасляну кислоту у вигляді твердої речовини (2,3 г). РХ/МС: RT=2,52 хв., МС: 464 (М+Н). Стадія 2. Методом, аналогічним описаному на стадії 5 методу А у прикладі 1(а), але використовуючи 4-{4-хлор-3-[2-(2,4дихлорфеніл)етилсульфамоїл)феніл}-4оксомасляну кислоту (0,83 г) замість 4-[4-хлор-3(2,4-дихлорбензил-сульфамоїл)феніл]-4оксомасляної кислоти, одержують (2-{4-хлор-3-[2(2,4-дихлорфеніл)етилсульфамоїл]феніл}-1Ніндол-3-іл)оцтову кислоту (150 мг). РХ/МС: RT=2,64 1 хв, МС: 537 (М+Н). H ЯМР (300 МГц, ДМСО-D6) 2,81 (т, J=7 Гц, 2Н), 3,2 (м, 2Н), 3,75 (с, 2Н), 7,06 (т, J=7,2 Гц, 1Н), 7,16 (т, J=7,3 Гц, 1Н), 7,29 (с, 2Н), 7,43 (м, 2Н), 7,56 (д, J=7,7 Гц, 1Н), 7,74 (д, J=8,2 Гц, 1Н), 7,9 (дд, J=6,3 Гц, J=2,2 Гц, 1Н), 8,13 (т, J=5,7 Гц, 1Н), 8,23 (д, J=2,2 Гц, 1Н), 11,52 (с, 1Н), 12,4 (уш.с, 1Н). IС50=2 нМ. Приклад 2 (а) (2-{4-Хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтова кислота Стадія 1. Суміш 2-хлорнітробензолу (53 г, 0,34 моль), заліза (1,5 г) і брому (23 мл, 0,45 моль) перемішують при нагріванні зі зворотним холодильником у потоці N2 протягом 20 годин. Реакційну суміш концентрують і залишок чистять флешхроматографією на силікагелі з елююванням 10% 95303 28 EtOAc-гептан. Потрібні фракції концентрують, фільтрують і промивають етанолом, а потім сушать. Проводять перекристалізацію твердого залишку з етанолу для одержання 5-бром-2хлорнітробензолу (37,9 г). Після зберігання маточних розчинів протягом ночі при 0°С виділяють другу порцію продукту і сушать її для одержання додаткової кількості 5-бром-2-хлорнітробензолу (7 г). МС: 235 (М+Н); т. пл. 65-67°С. Стадія 2. Розчин 5-бром-2-хлорнітробензолу (10,3 г, 43,6 ммоль) в EtOAc (200 мл) піддають гідрогенізації на нікелі Ренея (6 г 50% в Н2О) під 55 фунтів на кв. дюйм Н2 протягом 5 годин. Суміш фільтрують через шар целіту і промивають EtOAc. Фільтрат обробляють ефірною HCl (60 мл, 1М розчин в Еt2О) у потоці N2. Одержану суспензію перемішують протягом 1 години і додають Еt2О (100200 мл). Суміш фільтрують для одержання 5бром-2-хлоранілін гідрохлориду (4,85 г) у вигляді твердої речовини. МС: 205 (М+Н); т. пл. 152-155°С. Стадія 3. Суспензію 5-бром-2-хлоранілін гідрохлориду (41,4 г, 0,17 моль) в CH3CN (380 мл) охолоджують до 5°С і протягом 10 хвилин підливають концентровану HCl (277 мл). Суспензію охолоджують до -5°С і по краплях протягом 10-15 хвилин підливають розчин NaNO2 (14,2 г, 0,21 моль) в Н2О (40 мл). Суміш перемішують ще 5 хвилин і додають 30% (ваг./ваг.) SO2 в НОАс (435 мл) при 0°С, а потім підливають розчин дигідрату хлориду міді(ІІ) (15,3 г, 0,09 моль) в Н2О (40 мл). Реакційну суміш перемішують при кімнатній температурі протягом 1,5 години. Реакційну суміш фільтрують і твердий залишок сушать для одержання 5-бром-2хлорбензолсульфонілхлориду (18,4 г). Фільтрат зберігають при 0°С протягом 18 годин. Осад збирають і сушать для одержання додаткової кількості 5-бром-2-хлорбензолсульфонілхлориду (9,6 г). МС: 288 (М+Н). Стадія 4. 5-бром-2хлорбензолсульфонілхлорид (2 г, 6,9 ммоль) повільно додають у розчин 2-(2метоксифеніл)етиламіну (1,6 г, 10,74 ммоль) і DIEA (2,3 г, 17,8 ммоль) у DCM:MeOH (1:1, 50 мл) при 0°С. Одержану суміш нагрівають до кімнатної температури і перемішують протягом 20 годин. Реакційну суміш підкислюють водним 2н розчином HCl (~25 мл), а потім двічі екстрагують з DCM (~50 мл). Органічний шар промивають водою, сольовим розчином, сушать над сульфатом натрію і випарюють у вакуумі для одержання 5-бром-2-хлор-N[2-(2-метоксифеніл)етил]бензолсульфонаміду (2,23 г) у вигляді білого порошку. РХ/МС: RT=2,78 хв., МС: 403 (М+Н). Стадія 5. До розчину 1-(трет-бутоксикарбоніл)5-метоксі-1Н-індол-2-ілборонової кислоти (2,2 г, 7,5 ммоль), 5-бром-2-хлор-N-[2-(2-метоксифеніл)етил]бензолсульфонаміду (2 г, 5 ммоль) і CsF (1,14 г, 7,5 ммоль) у діоксан-Н2О (100 мл, 9:1) додають PdCl2(dppf)2 (400 мг) при кімнатній температурі у потоці N2. Реакційну суміш нагрівають до 80°С і перемішують протягом 2 годин. Реакційну суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і фільтрують через коротку колонку з силікагелем. Фільтрат концентрують у вакуумі і залишок очищають флеш-хроматографією на си 29 лікагелі, елююючи від 3% до 30% EtOAc у гептані для одержання трет-бутилового ефіру 2-{4-хлор-3[2-(2-метоксифеніл)етилсульфамоїл]феніл} індол1-карбонової кислоти (1,9 г). РХ/МС: RT=3,4 хв, МС: 541 (М+Н). Стадія 6. Суміш трет-бутилового ефіру 2-{4хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}індол-1карбонової кислоти (1,2 г, 2,2 ммоль) і TFA:DCM (1:1, 20 мл) перемішують протягом 3 годин при кімнатній температурі у вакуумі. Залишок розчиняють в EtOAc і промивають у насиченому водному розчині NaHCO3, воді і сольовому розчині. Органічну фазу відділяють, сушать над Na2SO4 і концентрують. Залишок очищають флешхроматографією на силікагелі, елююючи від 5% до 40% EtOAc у гептані для одержання 2-хлор-5-(1Ніндол-2-іл)-N-[2-(2метоксифеніл)етил]бензолсульфонаміду у вигляді порошку (980 мг). РХ/МС: RT=3 хв., МС: 441 (М+Н). Стадія 7. Оксалілхлорид (2М у DCM, 2 мл) повільно додають до розчину 2-хлор-5-(1Н-індол-2іл)-N-[2-(2метоксифеніл)етил]бензолсульфонаміду (600 мг, 1,36 ммоль) у DCM (15 мл) при 0°С. Реакційну суміш доводять до кімнатної температури. Після перемішування протягом 3 годин додають МеОН (5 мл) і перемішують протягом ще 10 хвилин. Суміш концентрують. Залишок очищають флешхроматографією на силікагелі, елююючи 10%-50% EtOAc у гептані для одержання метилового ефіру (2-{4-хлор-3-[2-(2-метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3-іл)оксооцтової кислоти у вигляді напівтвердої речовини (540 мг). РХ/МС: RT=2,72 хв., МС: 527 (М+Н). Стадія 8. Триетилсилан (0,5 мл) повільно додають до розчину метилового ефіру (2-{4-хлор-3[2-(2-метоксифеніл)етилсульфамоїл]феніл}-1Ніндол-3-іл)оксооцтової кислоти (500 мг, 0,95 ммоль) у TFA (5 мл) при кімнатній температурі. Після перемішування протягом 16 годин реакційну суміш концентрують у вакуумі. Залишок очищають флеш-хроматографією на силікагелі, елююючи 10%-40% EtOAc у гептані для одержання метилового ефіру (2-{4-хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти у вигляді напівтвердої речовини 1 (440 мг). РХ/МС: RT=2,88 хв., МС: 513 (М+Н); H ЯМР (300 МГц, CDCl3) 2,81 (т, J=6,8 Гц, 2Н), 3,23 (кв, J=12,6 Гц, J=6,6 Гц, 2Н), 3,72 (с, 3Н), 3,73 (с, 3Н), 3,81 (с, 2Н), 5,24 (т, J=5,7 Гц, 1Н), 6,82 (м, 2Н), 7,02 (д, J=7,3 Гц, 1Н), 7,2 (м, 3Н), 7,4 (д, J=7,9 Гц, 1Н), 7,53 (д, J=8,2 Гц, 1Н), 7,67 (д, J=7,7 Гц, 1Н), 7,86 (дд, J=6,1 Гц, 2,2 Гц, 1Н), 8,3 (д, J=2,2 Гц, 1Н), 8,5 (с, 1Н). Стадія 9. До розчину метилового ефіру (2-{4хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти (400 мг, 0,8 ммоль) у МеОН/Н2О (2:1, 20 мл) додають моногідрат гідроксиду літію (200 мг, 4,8 ммоль). Реакційну суміш перемішують при температурі 80°С протягом 3 годин і концентрують. Осад підкислюють водним 2н розчином HCl (рН ~2 мл), а потім двічі екстрагують з етилацетатом. Об'єднану органічну фазу промивають водою, 95303 30 сольовим розчином, сушать над сульфатом натрію і випарюють у вакуумі. Залишок очищають флешхроматографією на силікагелі, елююючи 20%-60% EtOAc у гептані для одержання (2-{4-хлор-3-[2-(2метоксифеніл)етил-сульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти у вигляді порошку (310 мг). 1 РХ/МС: RT=2,57 хв, МС: 497 (М-Н); H ЯМР (300 МГц, ДМСО-D6) 2,67 (т, J=8 Гц, 2H), 3,1 (м, 2Н), 3,62 (с, 3Н), 3,75 (с, 2Н), 6,82 (м, 2Н), 7,02-7,2 (м, 4Н), 7,41 (д, J=8,1 Гц, 1Н), 7,56 (д, J=7,9 Гц, 1Н), 7,78 (д, J=8,2 Гц, 1Н), 7,91 (дд, J=6,3 Гц, 2,2 Гц, 1Н), 8,01 (т, J=5,7 Гц, 1Н), 8,25 (д, J=2,2 Гц, 1Н), 11,55 (с, 1Н), 12,42 (с, 1Н). IС50=3,8 нМ. (b) (2-{4-Хлор-3-[2-(3метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтова кислота Стадія 1. Методом, аналогічним описаному на стадії 4 у прикладі 2(а), але використовуючи 2-(3метоксифеніл)етиламін замість 2-(2метоксифеніл)етиламіну, одержують 5-бром-2хлор-N-[2-(3метоксифеніл)етил]бензолсульфонамід у вигляді твердої речовини (2,2 г). РХ/МС: RT=2,71 хв., МС: 402 (М-Н). Стадія 2. Методом, аналогічним описаному на стадії 5 у прикладі 2(а), але використовуючи 5бром-2-хлор-N-[2-(3метоксифеніл)етил]бензолсульфонамід замість 5бром-2-хлор-N-[2-(2метоксифеніл)етил]бензолсульфонаміду, одержують трет-бутиловий ефір 2-{4-хлор-3-[2-(3метоксифеніл)етил-сульфамоїл] феніл} індол-1карбонової кислоти у вигляді масла (1,69 г). РХ/МС: RT=3,34 хв., МС: 541 (М+Н). Стадія 3. Методом, аналогічним описаному на стадії 6 у прикладі 2(а), але використовуючи третбутиловий ефір 2-{4-хлор-3-[2-(3метоксифеніл)етил-сульфамоїл]феніл}індол-1карбонової кислоти замість трет-бутилового ефіру 2-{4-хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}індол-1карбонової кислоти, одержують 2-хлор-5-(1Ніндол-2-іл)-N-[2-(3метоксифеніл)етил]бензолсульфонамід у вигляді твердої речовини (960 мг). РХ/МС: RT=2,92 хв., МС: 441 (М+Н). Стадія 4. Методом, аналогічним описаному на стадії 7 у прикладі 2(а), але використовуючи 2хлор-5-(1Н-індол-2-іл)-N-[2-(3метоксифеніл)етил]бензолсульфонамід замість 2хлор-5-(1Н-індол-2-іл)-N-[2-(2метоксифеніл)етил]бензолсульфонаміду, одержують метиловий ефір (2-{4-хлор-3-[2-(3метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оксооцтової кислоти у вигляді напівтвердої речовини (551 мг). РХ/МС: RT=2,66 хв., МС: 527 (М+Н). 31 Стадія 5. Методом, аналогічним описаному на стадії 8 у прикладі 2(а), але використовуючи метиловий ефір (2-{4-хлор-3-[2-(3-метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3-іл)оксооцтової кислоти замість метилового ефіру (2-{4-хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оксооцтової кислоти, одержують метиловий ефір (2-{4-хлор-3-[2-(3метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти (450 мг). РХ/МС: RT=2,82 хв., 1 МС: 513 (М+Н). H ЯМР (300 МГц, CDCl3) 2,78 (т, J=6,8 Гц, 2Н), 3,27 (кв, J=13 Гц, J=6,6 Гц, 2Н), 3,73 (с, 3Н), 3,75 (с, 3Н), 3,81 (с, 2Н), 5,07 (т, J=6 Гц, 1Н), 6,7 (м, 3Н), 7,2 (м, 3Н), 7,4 (д, J=8,1 Гц, 1Н), 7,55 (д, J=8,1 Гц, 1Н), 7,68 (д, J=7,9 Гц, 1Н), 7,86 (дд, J=6,1 Гц, 2,2 Гц, 1Н), 8,3 (д, J=2,2 Гц, 1Н), 8,47 (с, 1Н). Стадія 6. Методом, аналогічним описаному на стадії 9 у прикладі 2(а), але використовуючи метиловий ефір (2-{4-хлор-3-[2-(3-метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3-іл)оцтової кислоти замість метилового ефіру (2-{4-хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти, одержують (2-{4-хлор-3-[2-(3метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтову кислоту у вигляді твердої речовини (320 1 мг). РХ/МС: RT=2,5 хв., МС: 497 (М-Н). H ЯМР (300 МГц, ДМСО-D6) 2,69 (т, J=7,2 Гц, 2H), 3,18 (м, 2Н), 3,67 (с, 3Н), 3,75 (с, 2Н), 6,7 (т, J=6,5 Гц, 3Н), 7,12 (м, 3Н), 7,41 (д, J=8,1 Гц, 1Н), 7,57 (д, J=7,9 Гц, 1Н), 7,76 (д, J=8,2 Гц, 1Н), 7,9 (дд, J=6,2 Гц, 2,1 Гц, 1Н), 8,04 (уш.т, 1Н), 8,25 (д, J=2,1 Гц, 1Н), 11,55 (с, 1Н), 12,45 (уш.с, 1Н). IС50=3,3 нМ. (с) (2-{4-Хлор-3-[2-(4метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтова кислота Стадія 1. Методом, аналогічним описаному на стадії 4 у прикладі 2(а), але використовуючи 2-(4метоксифеніл)етиламін замість 2-(2метоксифеніл)етиламіну, одержують 5-бром-2хлор-N-[2-(4метоксифеніл)етил]бензолсульфонамід у вигляді напівтвердої речовини (2,3 г). РХ/МС: RT=2,71 хв., МС: 402 (М-Н). Стадія 2. Методом, аналогічним описаному на стадії 5 у прикладі 2(а), але використовуючи 5бром-2-хлор-N-[2-(4метоксифеніл)етил]бензолсульфонамід замість 5бром-2-хлор-N-[2-(2метоксифеніл)етил]бензолсульфонаміду, одержують трет-бутиловий ефір 2-{4-хлор-3-[2-(4метоксифеніл)етил-сульфамоїл]феніл}індол-1карбонової кислоти у вигляді масла (1,87 г). РХ/МС: Rt=3,33 хв., MC:541(M+H). Стадія 3. Методом, аналогічним описаному на стадії 6 у прикладі 2(а), але використовуючи третбутиловий ефір 2-{4-хлор-3-[2-(4метоксифеніл)етил-сульфамоїл]феніл}індол-1 95303 32 карбонової кислоти замість трет-бутилового ефіру 2-{4-хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}індол-1карбонової кислоти, одержують 2-хлор-5-(1Ніндол-2-іл)-N-[2-(4метоксифеніл)етил]бензолсульфонамід у вигляді твердої речовини (950 мг). РХ/МС: RT=2,91 хв., МС: 441 (М+Н). Стадія 4. Методом, аналогічним описаному на стадії 7 у прикладі 2(а), але використовуючи 2хлор-5-(1Н-індол-2-іл)-N-[2-(4метоксифеніл)етил]бензолсульфонамід замість 2хлор-5-(1Н-індол-2-іл)-N-[2-(2метоксифеніл)етил]бензолсульфонаміду, одержують метиловий ефір (2-{4-хлор-3-[2-(4метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оксооцтової кислоти у вигляді напівтвердої речовини (552 мг). РХ/МС: RT=2,66 хв., МС: 527 (М+Н). Стадія 5. Методом, аналогічним описаному на стадії 8 у прикладі 2(а), але використовуючи метиловий ефір (2-{4-хлор-3-[2-(4-метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3-іл)оксооцтової кислоти замість метилового ефіру (2-{4-хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оксооцтової кислоти, одержують метиловий ефір (2-{4-хлор-3-[2-(4метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти (445 мг). РХ/МС: RT=2,81 хв., 1 МС: 513 (М+Н); H ЯМР (300 МГц, CDCl3) 2,76 (т, J=6,6 Гц, 2Н), 3,23 (м, 2Н), 3,73 (с, 2Н), 3,77 (с, 3Н), 3,82 (с, 2Н), 5 (т, J=6,1 Гц, 1Н), 6,78 (дд, J=5 Гц, 2 Гц, 2Н), 7,02 (дд, J=6,6 Гц, 2 Гц, 2Н), 7,26 (м, 3Н), 7,41 (д, J=8,3 Гц, 1Н), 7,58 (д, J=8,3 Гц, 1Н), 7,69 (д, J=7,9 Гц, 1Н), 7,88 (дд, J=6,0 Гц, 2,2 Гц, 1Н), 8,3 (д, J=2,2Гц, 1Н), 8,33 (с, 1Н). Стадія 6. Методом, аналогічним описаному на стадії 9 у прикладі 2(а), але використовуючи метиловий ефір (2-{4-хлор-3-[2-(4-метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3-іл)оцтової кислоти замість метилового ефіру (2-{4-хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти, одержують (2-{4-хлор-3-[2-(4метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтову кислоту у вигляді твердої речовини (295 1 мг). РХ/МС: RT=2,51 хв., МС: 497 (М-Н). H ЯМР (300 МГц, ДМСО-D6) 2,64 (т, J=7,4 Гц, 2Н), 3,11 (м, 2Н), 3,67 (с, 3Н), 3,75 (с, 2Н), 6,75 (д, J=8,6 Гц, 2Н), 7,05 (м, 3Н), 7,17 (т, J=7,3 Гц, 1Н), 7,41 (д, J=8,1 Гц, 1Н), 7,58 (д, J=7,9 Гц, 1Н), 7,76 (д, J=8,2 Гц, 1Н), 7,9 (дд, J=6,3 Гц, 2 Гц, 1Н), 8,01 (уш.с, 1Н), 8,23 (д, J=2 Гц, 1Н), 11,54 (с, 1Н), 12,42 (уш.с, 1Н). IС50=3 нМ. (d) (2-{4-Хлор-3-[2-(2трифторметоксифеніл)етилсульфамоїл]феніл}-1Ніндол-3-іл)оцтова кислота Стадія 1. Методом, аналогічним описаному на стадії 4 у прикладі 2(а), але використовуючи 2-(2 33 трифторметоксифеніл)етиламін (2,2 г) замість 2(2-метоксифеніл)етиламіну, одержують 5-бром-2хлор-N-[2-(2-трифторметоксифеніл)етил]бензолсульфонамід у вигляді твердої речовини (2,4 г). РХ/МС: RT=2,96 хв, МС: 455,9 (МН). Стадія 2. Методом, аналогічним описаному на стадії 5 у прикладі 2(а), але використовуючи 5бром-2-хлор-N-[2-(2трифторметоксифенщ)етил]бензолсульфонамід (2,3 г) замість 5-бром-2-хлор-N-[2-(2метоксифеніл)етил]бензол-сульфонаміду, одержують трет-бутиловий ефір 2-{4-хлор-3-[2-(2трифторметоксифеніл)етилсульфамоїл]феніл}індол-1-карбонової кислоти у вигляді твердої речовини (2,05 г). РХ/МС: RT=3,49 хв., МС: 595 (М+Н). Стадія 3. Методом, аналогічним описаному на стадії 6 у прикладі 2(а), але використовуючи третбутиловий ефір 2-{4-хлор-3-[2-(2-трифторметоксифеніл)етилсульфамоїл]феніл}індол-1-карбонової кислоти замість трет-бутилового ефіру 2-{4-хлор3-[2-(2метоксифеніл)етилсульфамоїл]феніл}індол-1карбонової кислоти, одержують 2-хлор-5-(1Ніндол-2-іл)-N-[2-(2трифторметоксифеніл)етил]бензолсульфонамід у вигляді порошку (985 мг). РХ/МС: RT=3,1 хв, МС: 493 (М-Н). Стадія 4. Методом, аналогічним описаному на стадії 7 у прикладі 2(а), але використовуючи 2хлор-5-(1Н-індол-2-іл)-N-[2-(2трифторметоксифеніл)етил]бензолсульфонамід замість 2-хлор-5-(1Н-індол-2-іл)-N-[2-(2-метоксифеніл)етил]бензолсульфонаміду, одержують метиловий ефір (2-{4-хлор-3-[2-(2трифторметоксифеніл)етилсульфамоїл]феніл}-1Ніндол-3-іл)оксооцтової кислоти у вигляді твердої речовини (575 мг). РХ/МС: RT=2,84 хв., МС: 581 (М+Н). Стадія 5. Методом, аналогічним описаному на стадії 8 у прикладі 2(а), але використовуючи метиловий ефір (2-{4-хлор-3-[2-(2-трифторметоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оксооцтової кислоти замість метилового ефіру (2-{4-хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оксооцтової кислоти, одержують метиловий ефір (2-{4-хлор-3-[2-(2трифторметоксифеніл)етилсульфамоїл]феніл}-1Ніндол-3-іл)оцтової кислоти у вигляді порошку (470 1 мг). РХ/МС: RT=3 хв., МС: 567 (М+Н). H ЯМР (300 МГц, CDCl3) 2,91 (т, J=7 Гц, 2Н), 3,27 (кв, J=13,4 Гц, J=6,8 Гц, 2Н), 3,73 (с, 3Н), 3,81 (с, 2Н), 5,07 (т, J=6 Гц, 1Н), 7,25 (м, 6Н), 7,41 (д, J=7,9 Гц, 1Н), 7,59 (д, J=8,2 Гц, 1Н), 7,7 (д, J=7,59 Гц, 1Н), 7,91 (дд, J=6,1 Гц, 2,2 Гц, 1Н), 8,28 (с, 1Н), 8,33 (д, J=2,2 Гц, 1Н). Стадія 6. Методом, аналогічним описаному на стадії 9 у прикладі 2(а), але використовуючи метиловий ефір (2-{4-хлор-3-[2-(2трифторметоксифеніл)етилсульфамоїл]феніл}-1Ніндол-3-іл)оцтової кислоти замість метилового ефіру (2-{4-хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3 95303 34 іл)оцтової кислоти, одержують (2-{4-хлор-3-[2-(2трифторметоксифеніл)етилсульфамоїл]феніл}-1Ніндол-3-іл)оцтову кислоту у вигляді порошку (340 1 мг). РХ/МС: RT=2,69 хв., МС: 551 (М-Н). H ЯМР (300 МГц, ДМСО-D6) 2,8 (т, J=7 Гц, 2Н), 3,17 (кв, J=13,4 Гц, J=6,4 Гц, 2Н), 3,74 (с, 2Н), 7,06 (т, J=7,5 Гц, 1Н), 7,17 7,35 (м, 5Н), 7,41 (д, J=7,9 Гц, 1Н), 7,57 (д, J=7,9 Гц, 1Н), 7,79 (д, J=8,5 Гц, 2,1 Гц, 1Н), 7,91 (дд, 3=6,2 Гц, 2,1 Гц, 1Н), 8,17 (т, J=5,6 Гц, 1Н), 8,24 (д, J=2 Гц, 1Н), 11,57 (с, 1Н), 12,42 (уш.с, 1Н). IС50=20 нМ. (e) [2-(4-Хлор-3-фенетилсульфамоїлфеніл)1Н-індол-3-іл]оцтова кислота Стадія 1. Методом, аналогічним описаному на стадії 4 у прикладі 2(а), але використовуючи фенетиламін (1,3 г) замість 2-(2метоксифеніл)етиламіну, одержують 5-бром-2хлор-N-фенетилбензолсульфонамід (2,1 г) у вигляді твердої речовини. РХ/МС: RT=2,71 хв., МС: 402 (М-Н). Стадія 2. Методом, аналогічним описаному на стадії 5 у прикладі 2(а), але використовуючи 5бром-2-хлор-N-фенетилбензолсульфонамід (1,9 г) замість 5-бром-2-хлор-N-[2-(2метоксифеніл)етил]бензолсульфонаміду, одержують трет-бутиловий ефір 2-(4-хлор-3фенетилсульфамоїлфеніл)індол-1-карбонової кислоти (1,69 г). РХ/МС: RT=3,38 хв, МС: 511 (М+Н). Стадія 3. Методом, аналогічним описаному на стадії 6 у прикладі 2(а), але використовуючи третбутиловий ефір 2-(4-хлор-3фенетилсульфамоїлфеніл)індол-1-карбонової кислоти замість трет-бутилового ефіру 2-{4-хлор-3[2-(2-метоксифеніл)етилсульфамоїл]феніл}індол1-карбонової кислоти, одержують 2-хлор-5-(1Ніндол-2-іл)-N-фенетилбензолсульфонамід у вигляді твердої речовини (900 мг). РХ/МС: RT=2,96 хв., МС: 411 (М+Н). Стадія 4. Методом, аналогічним описаному на стадії 7 у прикладі 2(а), але використовуючи 2хлор-5-(1Н-індол-2-іл)-Nфенетилбензолсульфонамід замість 2-хлор-5-(1Ніндол-2-іл)-N-[2-(2метоксифеніл)етил]бензолсульфонаміду, одержують метиловий ефір (2-(4-хлор-3фенетилсульфамоїлфеніл)-1Н-індол-3іл)оксооцтової кислоти у вигляді напівтвердої речовини (542 мг). РХ/МС: RT=2,68 хв, МС: 497 (М+Н). Стадія 5. Методом, аналогічним описаному на стадії 8 у прикладі 2(а), але використовуючи метиловий ефір [2-(4-хлор-3фенетилсульфамоїлфеніл)-1Н-індол-3іл]оксооцтової кислоти замість метилового ефіру (2-{4-хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оксооцтової кислоти, одержують метиловий ефір [2-(4-хлор-3-фенетилсульфамоїлфеніл}-1Н 35 індол-3-іл)оцтової кислоти у вигляді твердої речовини (410 мг). РХ/МС: RT=2,84 хв, МС: 483 (М+Н); 1 H ЯМР (300 МГц, ДМСО-D6) 2,72 (т, J=7,5 Гц, 2Н), 3,15 (кв, J=13,4 Гц, J=6,5 Гц, 2Н), 3,61 (с, 3Н), 3,86 (с, 2Н), 7,15 (м, 7Н), 7,41 (д, J=8,1 Гц, 1Н), 7,55 (д, J=7,9 Гц, 1Н), 7,77 (д, J=8,3 Гц, 1Н), 7,87 (дд, J=6,2 Гц, 2 Гц, 1Н), 8,08 (т, J=5,5 Гц, 1Н), 8,2 (д, J=2 Гц, 1Н), 11,6 (с, 1Н). Стадія 6. Методом, аналогічним описаному на стадії 9 у прикладі 2(а), але використовуючи метиловий ефір [2-(4-хлор-3фенетилсульфамоїлфеніл)-1Н-індол-3-іл]оцтової кислоти замість метилового ефіру (2-{4-хлор-3-[2(2-метоксифеніл)етилсульфамоїл]феніл}-1Н-індол3-іл)оцтової кислоти, одержують [2-(4-хлор-3фенетилсульфамоїлфеніл)-1Н-індол-3-іл]оцтову кислоту (280 мг). РХ/МС: RT=2,53 хв., МС: 467 (М1 Н); H ЯМР (300 МГц, ДМСО-D6) 2,72 (т, J=7,4Гц, 2Н), 3,15 (м, 2Н), 3,75 (с, 2Н), 7,15 (м, 7Н), 7,41 (д, J=7,8 Гц, 1Н), 7,57 (д, J=7,8 Гц, 1Н), 7,77 (д, J=8,2 Гц, 1Н), 7,89 (дд, J=6,3 Гц, 2 Гц, 1Н), 8,06 (т, J=5,7 Гц, 1Н), 8,24 (д, J=2 Гц, 1Н), 11,55 (с, 1Н), 12,45 (уш.с, 1Н). (f) {2-[4-Хлор-3-(3фенілпропілсульфамоїл)феніл]-1Н-індол-3іл}оцтова кислота Стадія 1. Методом, аналогічним описаному на стадії 4 у прикладі 1, але використовуючи 3фенілпропіламін (1,4 г) замість 2-(2метоксифеніл)етиламіну, одержують 5-бром-2хлор-N-(3-фенілпропіл)бензолсульфонамід (2 г) у вигляді напівтвердої речовини. РХ/МС: RT=2,86 хв., МС: 386 (М-Н). Стадія 2. Методом, аналогічним описаному на стадії 5 у прикладі 2(а), але використовуючи бром2-хлор-N-(3-фенілпропіл)бензолсульфонамід (1,9 г) замість 5-бром-2-хлор-N-[2-(2метоксифеніл)етил]бензолсульфонаміду, одержують трет-бутиловий ефір 2-[4-хлор-3-(3фенілпропілсульфамоїл)феніл]індол-1-карбонової кислоти у вигляді твердої речовини (1,73 г). РХ/МС: RT=3,43 хв., МС: 525 (М+Н). Стадія 3. Методом, аналогічним описаному на стадії 6 у прикладі 2(а), але використовуючи третбутиловий ефір 2-[4-хлор-3-(3фенілпропілсульфамоїл)феніл]індол-1-карбонової кислоти замість трет-бутилового ефіру 2-{4-хлор3-[2-(2метоксифеніл)етилсульфамоїл]феніл}індол-1карбонової кислоти, одержують 2-хлор-5-(1Ніндол-2-іл)-N-(3-фенілпропіл)бензолсульфонамід у вигляді порошку (950 мг). РХ/МС: RT=3,03 хв., МС: 425 (М+Н). Стадія 4. Методом, аналогічним описаному на стадії 7 у прикладі 2(а), але використовуючи 2хлор-5-(1Н-індол-2-іл)-N-(3фенілпропіл)бензолсульфонамід замість 2-хлор-5(1Н-індол-2-іл)-N-[2-(2 95303 36 метоксифеніл)етил]бензолсульфонаміду, одержують метиловий ефір {2-[4-хлор-3-(3фенілпропілсульфамоїл)феніл]-1Н-індол-3іл}оксооцтової кислоти у вигляді напівтвердої речовини (540 мг). РХ/МС: Rt=2,76 хв.,MC: 511 (M+H). Стадія 5. Методом, аналогічним описаному на стадії 8 у прикладі 2(а), але використовуючи метиловий ефір {2-[4-хлор-3-(3фенілпропілсульфамоїл)феніл]-1Н-індол-3іл}оксооцтової кислоти замість метилового ефіру (2-{4-хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оксооцтової кислоти, одержують метиловий ефір {2-[4-хлор-3-(3фенілпропілсульфамоїл)феніл]-1Н-індол-3іл}оцтової кислоти (430 мг). РХ/МС: RT=2,92 хв., 1 МС: 497 (М+Н); H ЯМР (300 МГц, CDCl3) 1,82 (м, 2Н), 2,62 (т, J=7,5 Гц, 2Н), 3 (м, 2Н), 3,72 (с, 3Н), 3,8 (с, 2Н), 5,13 (т, J=6 Гц, 1Н), 7,07-7,28 (м, 7Н), 7,39 (д, J=8,1 Гц, 1Н), 7,6 (д, J=8,3 Гц, 1Н), 7,68 (д, J=7,9 Гц, 1Н), 7,89 (дд, J=6,1 Гц, 2,2 Гц, 1Н), 8,08 (т, 5,5 Гц, 1Н), 8,31 (д, J=2 Гц, 1Н). Стадія 6. Методом, аналогічним описаному на стадії 9 у прикладі 2(а), але використовуючи метиловий ефір {2-[4-хлор-3-(3фенілпропілсульфамоїл)феніл]-1Н-індол-3іл}оцтової кислоти замість метилового ефіру (2-{4хлор-3-[2-(2метоксифеніл)етилсульфамоїл]феніл}-1Н-індол-3іл)оцтової кислоти, одержують {2-[4-хлор-3-(3фенілпропілсульфамоїл)феніл]-1Н-індол-3іл}оцтову кислоту у вигляді порошку (300 мг). 1 РХ/МС: RT=2,61 хв., МС: 481 (М-Н); H ЯМР (300 МГц, ДМСО-D6) 1,67 (м, 2Н), 2,5 (м, 2Н, приховане піком ДМСО), 2,93 (м, 2Н), 3,73 (с, 2Н), 7 7,3 (м, 7Н), 7,41 (д, J=8,1 Гц, 1Н), 7,57 (д, J=7,8 Гц, 1Н), 7,83 (д, J=8,4 Гц, 1Н), 7,93 (д, J=8,1 Гц, 1Н), 8,04 (очевидно с, 1Н), 8,26 (с, 1Н), 11,6 (с, 1Н), 12,45 (уш.с, 1Н). IС50=7 нМ. Приклад 3 2-[2-(4-Хлор-3-циклогексилсульфамоїлфеніл)1Н-індол-3-іл]-N-метилацетамід Стадія 1. 5-бром-2хлорбензолсульфонілхлорид (4 г, 13,8 ммоль) повільно додають до розчину циклогексиламіну (3,5 г, 35 ммоль) у DCM:MeOH (1:1, 100 мл) при температурі 0°С. Одержану суміш нагрівають до кімнатної температури і перемішують протягом 20 годин. Реакційну суміш підкислюють водним 2 N розчином HCl (~100 мл), а потім двічі екстрагують DCM (~150 мл). Органічний шар промивають водою (~100 мл), насиченим сольовим розчином (~50 мл), сушать над сульфатом натрію і випарюють у вакуумі для одержання 5-бром-2-хлор-Nциклогексилбензолсульфонаміду (4,2 г). РХ/МС: RT=3 хв., МС: 351 (М-Н). 37 Стадія 2. До розчину 1-(трет-бутоксикарбоніл)1Н-індол-2-ілборонової кислоти (2,2 г), 5-бром-2хлор-N-циклогексилбензолсульфонаміду (1,8 г) і CsF (1,4 г) в 1,4-діоксан-Н2О (60 мл, 10:1) додають PdCl2(dppf)2 (375 мг) при кімнатній температурі у потоці азоту. Реакційну суміш нагрівають до 80°С і перемішують протягом 3 годин. Реакційну суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і фільтрують через коротку колонку з силікагелем. Фільтрат концентрують у вакуумі і очищають флеш-хроматографією на силікагелі, елююючи від 5% до 50% EtOAc у гептані для одержання трет-бутилового ефіру 2-(4-хлор-3циклогексилсульфамоїлфеніл)індол-1-карбонової кислоти (1,9 г). РХ/МС: RT=3,31 хв., МС: 489 (М+Н). Стадія 3. Суміш трифтороцтової кислоти (10 мл) і дихлорметану (10 мл) додають до третбутилового ефіру 2-(4-хлор-3циклогексилсульфамоїлфеніл)індол-1-карбонової кислоти (1,9 г). Реакційну суміш перемішують при кімнатній температурі протягом 2 годин. Суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають у насиченому водному розчині NaHCO3, воді і сольовому розчині. Органічну фазу відділяють, сушать над сульфатом натрію і концентрують для одержання 2-хлор-N-циклогексил-5(5-метоксі-1Н-індол-2-іл)бензолсульфонаміду (1,4 г). РХ/МС: RT=3,17 хв., МС: 389 (М+Н). Стадія 4. Оксалілхлорид (1,7 мл 2М розчину у дихлорметані) повільно додають до розчину 2хлор-N-циклогексил-5-(1Н-індол-2іл)бензолсульфонаміду (300 мг, 0,77 ммоль) у DCM (6 мл) при температурі 0°С. Реакційну суміш доводять до кімнатної температури і перемішують протягом 3 годин. Додають метиламін у THF (7 мл 2М розчину) і перемішують протягом 15 хв. Суміш концентрують і залишок очищають методом хроматографії на силікагелі, елююючи 30-70% EtOAc/гептану для одержання 2-[2-(4-хлор-3-цикл огексилсульфамоїлфеніл)-1Н-індол-3-іл]-N-метил2-оксоацетаміду у вигляді порошку (285 мг). 1 РХ/МС: RT=2,22 хв., МС: 474 (М+Н); H ЯМР (300 МГц, ДМСО-D6) 0,9-1,7 (серії м, 10Н), 2,36 (д, J=4,7 Гц, 3Н), 3,02 (м, 1Н), 7,3 (м, 2Н), 7,52 (д, J=8 Гц, 1Н), 7,76 (м, 2Н), 7,99 (д, J=8 Гц, 1Н), 8,06 (дд, J=5 Гц, J=1,8 Гц, 1Н), 8,15 (д, J=1,8 Гц, 1Н), 8,49 (д, J=4,8 Гц, 1Н), 12,65 (с, 1Н). Стадія 5. Триетилсилан (1 мл) повільно додають до розчину 2-[2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3-іл]-Nметил-2-оксоацетаміду (150 мг; 0,32 ммоль) у TFA (4 мл) при кімнатній температурі. Після перемішування протягом -72 годин реакційну суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають у насиченому водному розчині NaHCO3, воді, сушать над Na2SO4 і концентрують. Залишок очищають препаративним розділенням ВЕРХ (рухома фаза: ацетонітрил-вода з 0,1% TFA; градієнт 10-100% протягом 10 хв.) для одержання 2-[2-(4-хлор-3-циклогексидсульфамоїлфеніл)-1Ніндол-3-іл]-N-метилацетаміду у вигляді напівтвердої речовини (110 мг). РХ/МС: RT=2,6 хв., МС: 460 1 (М+Н); Н ЯМР (300 МГц, ДМСО-D6) 0,9-1,7 (м, 10Н), 2,6 (д, J=4,6 Гц, 3Н), 3,04 (м, 1Н), 3,6 (с, 2Н), 7,03 (т, J=7,4 Гц, 1Н), 7,16 (т, J=7,4 Гц, 1Н), 7,4 (д, 95303 38 J=8 Гц, 1Н), 7,6 (д, J=7,9 Гц, 1Н), 7,76 (д, J=8,4 Гц, 1Н), 7,89 (д, J=8,3 Гц, 1Н), 8,01 (д, J=4,6 Гц, 1Н), 8,14 (дд, J=6 Гц, J=2,2 Гц, 1Н), 8,33 (д, J=2 Гц, 1Н), 11,5 (с, 1Н). IC50=509 нМ. Приклад 4 [4-Хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтова кислота Метод А: Стадія 1. Ди-трет-бутилдикарбонат (39,6 г) додають до розчину 4-хлоріндолу (25 г) і 4(диметиламіно)піридину (2 г) у DCM (800 мл). Реакційну суміш перемішують при кімнатній температурі протягом 18 годин. Реакційну суміш промивають 1н HCl (150 мл) і 1н NaHCO3 (150 мл). Органічну фазу відділяють, сушать над MgSO4 і концентрують. Сирий залишок перекристалізовують з гептану/ефіру для одержання третбутилового ефіру 4-хлоріндол-1-карбонової кислоти (41,9 г). РХ/МС: RT=3,34 хв., МС: 251 (М+Н). Стадія 2. До розчину трет-бутилового ефіру 4хлоріндол-1-карбонової кислоти (10 г) у сухому THF (50 мл) додають триізопропілборат (13,7 мл) у потоці N2. Суміш охолоджують до 0°С у льодяній бані. Літійдіізопропіламін (33,8 мл, 2М) додають протягом години при температурі 0°С. Реакційну суміш перемішують при температурі 0°С протягом 30 хв. Додають 2н HCl (80 мл). Одержану суміш екстрагують EtOAc. Органічну фазу сушать, фільтрують і концентрують. Залишок перекристалізовують в ацетонітрилі/Н2О для одержання 1-(третбутоксикарбоніл)-4-хлор-1Н-індол-2-ілборонової кислоти у вигляді твердої речовини (4,5 г). Стадія 3. До розчину 1-(трет-бутоксикарбоніл)4-хлор-1Н-індол-2-ілборонової кислоти (4,27 г, 14,45 ммоль), 5-бром-2-хлор-Nциклогексилбензолсульфонаміду (3 г, 8,5 ммоль) і CsF (2,58 г, 17 ммоль) у діоксан-Н2О (85 мл, 10:1) додають PdCl2(dppf)2 (694 мг, 0,85 ммоль) при кімнатній температурі у потоці N2. Реакційну суміш нагрівають до 80°С і перемішують протягом 2 годин. Реакційну суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і фільтрують через коротку колонку з силікагелем. Фільтрат концентрують у вакуумі і залишок очищають флешхроматографією на силікагелі, елююючи від 5% до 50% EtOAc у гептані для одержання третбутилового ефіру 4-хлор-2-(4-хлор-3циклогексилсульфамоїл-феніл)індол-1-карбонової кислоти у вигляді твердої речовини (3,42 г). РХ/МС: RT=3,5 хв., МС: 523 (М+Н). Стадія 4. TFA (20 мл) додають до розчину трет-бутилового ефіру 4-хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)індол-1-карбонової кислоти (3,42 г, 6,53 ммоль) у DCM (40 мл). Реакційну суміш перемішують при кімнатній температурі протягом ночі. Суміш концентрують у вакуумі. 39 Залишок розчиняють в EtOAc і промивають 1н NaHCO3. Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання 2-хлор-5-(4хлор-1Н-індол-2-іл)-N-циклогексилбензолсульфонаміду у вигляді твердої речовини (2,8 г). РХ/МС: RT=3,04 хв., МС: 423 (М+Н). Стадія 5. Етилоксалілхлорид (2,42 г, 17,8 ммоль) повільно додають до суспензії 2-хлор-Nциклогексил-5-(1Н-індол-2-іл)бензолсульфонаміду (1,5 г, 3,54 ммоль) у дихлоретані (150 мл) з подальшим додаванням АlСl3 (2,36 г, 17,8 ммоль) при температурі 0°С. Одержаний темно-коричневий розчин доводять до кімнатної температури і перемішують протягом 16 годин. До реакційної суміші додають МеОН (5 мл) при температурі 0°С і розбавляють DCM. Органічну фазу промивають водою, сольовим розчином, сушать над Na2SO4 і концентрують для одержання етилового ефіру [4хлор-2-(4-хлор-3-циклогексилсульфамоїлфеніл)1Н-індол-3-іл]оксооцтової кислоти у вигляді твердої речовини (1,8 г). РХ/МС: RT=2,87 хв, МС: 523 1 (М+Н). H ЯМР (300 МГц, ДМСО-D6) 0,8-1,7 (серії м, 13Н), 3,04 (м, 1Н), 4,07 (кв, J=14,3 Гц, J=7,2 Гц, 2Н), 7,3 (м, 2Н), 7,54 (дд, J=4,2 Гц, 2,4 Гц, 1Н), 7,83 (м, 2Н), 8,04 (д, J=8,1 Гц, 1Н), 8,17 (д, J=2 Гц, 1Н), 12,95 (с, 1Н). Стадія 6. Етиловий ефір [4-хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оксооцтової кислоти (1,8 г, 3,45 ммоль) перемішують з триетилсиланом (6 мл) і TFA (24 мл) при кімнатній температурі. Після перемішування протягом ~72 годин реакційну суміш концентрують у вакуумі. Залишок розчиняють у DCM (~150 мл) і промивають двічі водою (~100 мл), потім сольовим розчином (~50 мл), сушать над Na2SO4 і концентрують у вакуумі. Залишок очищають флешхроматографією на силікагелі, елююючи 5%-50% EtOAc у гептані для одержання етилового ефіру [4-хлор-2-(4-хлор-3-циклогексилсульфамоїлфеніл)1Н-індол-3-іл]оцтової кислоти у вигляді порошку 1 (1,45 г). РХ/МС: RT=3,14 хв., МС: 509 (М+Н). Н ЯМР (300 МГц, ДМСО-D6) 0,8-1,7 (серії м, 13Н), 3,03 (м, 1H), 3,96 (с, 2Н), 4,14 (кв, J=14,2 Гц, J=7,2 Гц, 2Н), 7,07 (д, J=7,5 Гц, 1Н), 7,14 (т, J=7,8 Гц, 1Н), 7,4 (д, J=8 Гц, 1Н), 7,8 (м, 2Н), 7,98 (д, J=8,1 Гц, 1Н), 8,14 (д, J=2 Гц, 1Н), 11,95 (с, 1Н). Стадія 7. Суміш етилового ефіру [4-хлор-2-(4хлор-3-циклогексил-сульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти (1,45 г, 2,85 ммоль) і моногідрату гідроксиду літію (600 мг, 14,3 ммоль) у МеОН/Н2О (2:1, 100 мл) перемішують при температурі 80°С протягом 4 годин. До суміші додають КОН (800 мг; 14,3 ммоль) і продовжують перемішувати при температурі 80°С протягом 16 годин. Реакційну суміш концентрують у вакуумі. Залишок підкислюють водним 2н розчином HCl (рН ~2). Утворені білі тверді речовини були зібрані методом фільтрації, промиті Et2O, гептаном і висушені у вакуумі протягом ~72 годин для одержання [4-хлор-2-(4хлор-3-циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти у вигляді твердої кристалічної речовини (1,1 г). РХ/МС: Rt=2,64 хв., МС: 481 1 (М+Н); H ЯМР (300 МГц, ДМСО-D6) 0,9-1,7 (серії м, 10Н), 3,05 (м, 1Н), 3,88 (с, 2Н), 7,06 (д, J=7,4 Гц, 1Н), 7,14 (т, J=7,8 Гц, 1Н), 7,40 (д, J=7,9 Гц, 1Н), 95303 40 7,8 (м, 2Н), 7,97 (д, J=8,1 Гц, 1Н), 8,18 (д, J=1,8 Гц, 1Н), 11,92 (с, 1Н), 12,45 (уш.с, 1Н). IC50=0,2 нМ. Метод В: Стадія 1. Ди-трет-бутилдикарбонат (39,6 г) додають до розчину 4-хлоріндолу (25 г) і 4(диметиламіно)піридину (2 г) у DCM (800 мл). Реакційну суміш перемішують при кімнатній температурі протягом 18 годин. Реакційну суміш промивають 1н HCl (150 мл) і 1н NaHCO3 (150 мл). Органічну фазу відділяють, сушать над MgSO4 і концентрують. Сирий залишок перекристалізовують з гептану/ефіру для одержання третбутилового ефіру 4-хлоріндол-1-карбонової кислоти (41,9 г). Стадія 2. До розчину трет-бутилового ефіру 4хлоріндол-1-карбонової кислоти (10 г) у сухому THF (50 мл) додають триізопропілборат (13,7 мл) у потоці N2. Суміш охолоджують до 0°С у льодяній бані. Літійдіізопропіламін (33,8 мл, 2М) додають протягом години при температурі 0°С. Реакційну суміш перемішують при температурі 0°С протягом 30 хв. Додають 2н HCl (80 мл). Одержану суміш екстрагують EtOAc. Органічну фазу сушать, фільтрують і концентрують. Залишок перекристалізовують в ацетонітрилі/Н2О для одержання 1-(третбутоксикарбоніл)-4-хлор-1Н-індол-2-ілборонової кислоти у вигляді твердої речовини (4,5 г). Стадія 3. До розчину 1-(трет-бутоксикарбоніл)4-хлор-1Н-індол-2-ілборонової кислоти (1,04 г), 5бром-2-хлор-N-циклогексилбензолсульфонаміду (1 г) і CsF (864 мг) у діоксан-Н2О (29 мл, 10:1) додають PdCl2(dppf)2 (232 мг) при кімнатній температурі у присутності азоту. Реакційну суміш нагрівають до 80°С і перемішують протягом ночі. Реакційну суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і фільтрують через коротку колонку з силікагелем. Фільтрат концентрують у вакуумі і очищають флеш-хроматографією на силікагелі, елююючи 10%-50% EtOAc у гептані для одержання трет-бутилового ефіру 4-хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)індол-1-карбонової кислоти у вигляді твердої речовини (1,04 г). Стадія 4. Трифтороцтову кислоту (5 мл) додають до розчину трет-бутилового ефіру 4-хлор-2-(4хлор-3-циклогексилсульфамоїлфеніл)індол-1карбонової кислоти (1,04 г) у DCM (10 мл). Реакційну суміш перемішують при кімнатній температурі протягом 4 годин. Суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають 1н NaHCO3. Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання 2-хлор-5-(4хлор-1Н-індол-2-іл)-Nциклогексилбензолсульфонаміду у вигляді твердої речовини (860 мг). РХ/МС: RT=3,06 хв., МС: 423 (М+Н). Стадія 5. Оксалілхлорид (0,26 мл) повільно додають до розчину 2-хлор-5-(4-хлор-1Н-індол-2іл)-N-циклогексилбензолсульфонаміду (860 мг) у дихлорметані (20 мл) при кімнатній температурі. Після перемішування протягом 2 годин додають МеОН (5 мл) і перемішують протягом 15 хв. Суміш концентрують. Залишок очищають флешхроматографією на силікагелі, елююючи 10%-50% EtOAc у гептані для одержання метилового ефіру [4-хлор-2-(4-хлор-3-циклогексилсульфамоїлфеніл) 41 1Н-індол-3-іл]оксооцтової кислоти у вигляді твердої речовини (140 мг). Стадія 6. Триетилсилан (0,086 мл) повільно додають до розчину метилового ефіру [4-хлор-2(4-хлор-3-циклогексилсульфамоїлфеніл)-1Н-індол3-іл]оксооцтової кислоти (140 мг) у трифтороцтовій кислоті (1,4 мл) при кімнатній температурі. Після перемішування протягом ночі леткі фракції видаляють під вакуумом. Залишок розчиняють в EtOAc і промивають 1н NaHCO3. Органічну фазу відділяють, сушать над MgSO4 і концентрують. Залишок очищають флеш-хроматографією на силікагелі, елююючи 10%-50% EtOAc у гептані для одержання метилового ефіру [4-хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1H-індол-3іл]оцтової кислоти у вигляді твердої речовини (93 мг). Стадія 7. До розчину метилового ефіру [4хлор-2-(4-хлор-3-циклогексилсульфамоїлфеніл)1Н-індол-3-іл]оцтової кислоти (92 мг) у МеОН/Н2О (1:1, 3,6 мл) додають моногідрат гідроксиду літію (16 мг). Реакційну суміш перемішують при температурі 80°С протягом 18 годин. Додають EtOAc (10 мл) і розчин промивають 1н HCl (5 мл). Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання [4-хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти у вигляді твердої речовини (67 1 мг). РХ/МС: Rt=2,52 хв., МС: 481 (М+Н); H ЯМР (300 МГц, CD3OD) 1,09-1,35 (м, 5Н), 1,51-1,74 (м, 5Н), 3,11 (м, 1Н), 3,81 (уш.с, 2Н), 7,05 (м, 2Н), 7,39 (м, 1Н), 7,65 (м, 2Н), 8,32 (м,1Н), 11,17 (уш.с, 1Н). Приклад 5 Калій [4-хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]ацетат Суміш [4-хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти (537 мг, 1,115 ммоль) і 200 мл етанолу перемішують при температурі ~40°С протягом 10 хв. Одержаний розчин охолоджують до кімнатної температури і додають гідроксид калію (62 мг, 1,1 ммоль). Продовжують перемішування при кімнатній температурі до повного розчинення КОН. Розчин концентрують у вакуумі при температурі ~40°С. Утворені білі тверді речовини сушать у вакуумі протягом ~20 годин для одержання [4хлор-2-(4-хлор-3-циклогексилсульфамоїлфеніл)1Н-індол-3-іл]ацетату калію у вигляді твердої кристалічної речовини (575 мг). РХ/МС: RT=2,64 хв., 1 МС: 481 (М+Н вихідної кислоти). H ЯМР (300МГц, ДМСО-D6) 0,9-1,7 (серії м, 10Н), 3,06 (м, 1Н), 3,66 (с, 2Н), 6,93 (д, J=7,2 Гц, 1Н), 7,01 (т, J=7,8 Гц, 1Н), 7,28 (д, J=7,7 Гц, 1Н), 7,66 (д, J=8,3 Гц, 1Н), 8,1 95303 42 (дд, J=6,4 Гц, 2 Гц, 2Н), 8,32 (д, J=2 Гц, 1Н), 11,75 (с, 1Н). Приклад 6 [2-(4-Хлор-3-циклогексилсульфамоїлфеніл)-4фтор-1Н-індол-3-іл]оцтова кислота Стадія 1. Ди-трет-бутилдикарбонат (8,88 г) додають до розчину 4-фторіндолу (5 г) і 4(диметиламіно)піридину (0,45 г) у дихлорметані (185 мл). Реакційну суміш перемішують при кімнатній температурі протягом 4 годин. Реакційну суміш промивають 1н HCl (100 мл) і 1н NHCO3 (100 мл). Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання третбутилового ефіру 4-фторіндол-1-карбонової кислоти у вигляді масла (8,32 г). РХ/МС: RT=3,34 хв., МС: 236,09 (М+Н). Стадія 2. До розчину трет-бутилового ефіру 4фтор-індол-1-карбонової кислоти (3 г) у сухому THF (16 мл) додають триізопропілборат (3,6 мл) у присутності азоту. Суміш охолоджують до 0°С у льодяній бані. Літійдіізопропіламін (12,8 мл, 2М) додають протягом години при температурі 0°С. Реакційну суміш перемішують при температурі 0°С протягом 30 хв. Додають 2н HCl (10 мл), щоб погасити реакцію. Одержану суміш екстрагують EtOAc. Залишок очищають флеш-хроматографією на силікагелі, елююючи 5%-5О% EtOAc у гептані для одержання 1-(трет-бутоксикарбоніл)-4-фтор-1Ніндол-2-ілборонової кислоти у вигляді твердої речовини (1,65 г). Стадія 3. До розчину 1-(трет-бутоксикарбоніл)4-фтор-1Н-індол-2-ілборонової кислоти (1,19 г), 5бром-2-хлор-N-циклогексилбензолсульфонаміду (1 г) і CsF (863 мг) у діоксан-Н2О (27,5 мл, 10:1) додають PdCl2(dppf)2 (231 мг) при кімнатній температурі у присутності азоту. Реакційну суміш нагрівають до 80°С і перемішують протягом 2 днів. Реакційну суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і фільтрують через коротку колонку з силікагелем. Фільтрат концентрують у вакуумі і очищають флеш-хроматографією на силікагелі, елююючи 5%-30% EtOAc у гептані для одержання 2-хлор-5-(4-фтор-1Н-індол-2-іл)-Nциклогексилбензолсульфонаміду у вигляді твердої речовини (516 мг). РХ/МС: RT=4,46 хв, МС: 407 (М+Н). Стадія 4. Оксалілхлорид (0,16 мл) повільно додають до розчину 2-хлор-5-(4-фтор-1Н-індол-2іл)-N-циклогексилбензолсульфонаміду (496 мг) у дихлорметані (12 мл) при кімнатній температурі. Після перемішування протягом 3 годин додають МеОН (4 мл) і перемішують протягом 15 хв. Суміш концентрують. Залишок очищають флешхроматографією на силікагелі, елююючи 5%-50% EtOAc у гептані для одержання метилового ефіру [4-фтор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3 43 іл]оксооцтової кислоти у вигляді твердої речовини (470 мг). Стадія 5. Триетилсилан (0,3 мл) повільно додають до розчину метилового ефіру [4-фтор-2-(4хлор-3-циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оксооцтової кислоти (570 мг) у трифтороцтовій кислоті (5 мл) при кімнатній температурі. Після перемішування протягом ночі леткі фракції видаляють під вакуумом. Залишок розчиняють в EtOAc і промивають 1н NaHCO3. Органічну фазу відділяють, сушать над MgSO4 і концентрують. Залишок очищають флеш-хроматографією на силікагелі, елююючи 10%-50% EtOAc у гептані для одержання метилового ефіру [4-фтор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти у вигляді білої твердої речовини (350 мг). РХ/МС: RT=3,18 хв., МС: 479,1 (М+Н). Стадія 6. До розчину метилового ефіру [4фтор-2-(4-хлор-3-циклогексил-сульфамоїлфеніл)1Н-індол-3-іл]оцтової кислоти (250 мг) у МеОН/Н2О (1:1, 7 мл) додають моногідрат гідроксиду літію (44 мг). Реакційну суміш перемішують при температурі 80°С протягом ночі. Додають EtOAc (15 мл) і розчин промивають 1н HCl (10 мл). Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання [2-(4-хлор-3циклогексилсульфамоїлфеніл)-4-фтор-1Н-індол-3іл]оцтової кислоти у вигляді твердої речовини (219 1 мг). РХ/МС: RT=2,83 хв., МС: 465 (М+Н); H ЯМР (300 МГц, ДМСО) 1,09-1,24 (м, 5Н), 1,49-1,61 (м, 5Н), 3,07 (м, 1Н), 3,81 (с, 2Н), 6,8 (м, 1Н), 7,15 (м, 1Н), 7,26 (м, 1Н), 7,84 (м, 2Н), 7,98 (м, 1Н), 8,23 (м, 1Н), 11,86 (уш.с, 1Н). ІС50=0,7 нМ. Приклад 7 [2-(4-Хлор-3-циклогексилсульфамоїлфеніл)-4метил-1Н-індол-3-іл]оцтова кислота Стадія 1. Ди-трет-бутилдикарбонат (9,15 г) додають до розчину 4-метиліндолу (5 г) і 4(диметиламіно)піридину (0,46 г) у дихлорметані (190 мл). Реакційну суміш перемішують при кімнатній температурі протягом 4 годин. Реакційну суміш промивають 1н HCl (100 мл) і 1н NHCO3 (100 мл). Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання третбутилового ефіру 4-метиліндол-1-карбонової кислоти у вигляді масла (8,75 г). Стадія 2. До розчину трет-бутилового ефіру 4метиліндол-1-карбонової кислоти (3 г) у сухому THF (16 мл) додають триізопропілборат (4,45 мл) у присутності азоту. Суміш охолоджують до 0°С у льодяній бані. Додають літійдіізопропіламін (11,6 мл, 2М) протягом години при температурі 0°С. Реакційну суміш перемішують при температурі 0°С протягом 30 хв. Додають 2N HCl (10 мл), щоб погасити реакцію. Одержану суміш екстрагують EtOAc. Залишок перекристалізовують у CH3CN/H2O для одержання 1-(трет 95303 44 бутоксикарбоніл)-4-метил-1Н-індол-2-ілборонової кислоти у вигляді твердої речовини (1,53 г). Стадія 3. До розчину 1-(трет-бутоксикарбоніл)4-метил-1Н-індол-2-ілборонової кислоти (1,41 г), 5бром-2-хлор-N-циклогексилбензолсульфонаміду (1 г) і CsF (863 мг) у діоксан-Н2О (27,5 мл, 10:1) додають PdCl2(dppf)2 (232 мг)при кімнатній температурі у присутності азоту. Реакційну суміш нагрівають до 80°С і перемішують протягом ночі. Реакційну суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і фільтрують через коротку колонку з силікагелем. Фільтрат концентрують у вакуумі і очищають флеш-хроматографією на силікагелі, елююючи 0%-50% EtOAc у гептані для одержання трет-бутилового ефіру 2-(4-хлор-3циклогексилсульфамоїлфеніл)-4-метиліндол-1карбонової кислоти у вигляді твердої речовини (845 мг). Стадія 4. Трифтороцтову кислоту (5 мл) додають до розчину трет-бутилового ефіру 2-(4-хлор-3циклогексилсульфамоїлфеніл)-4-метиліндол-1карбонової кислоти (845 мг) у дихлорметані (10 мл). Реакційну суміш перемішують при кімнатній температурі протягом ночі. Суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають 1н NaHCO3. Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання 2хлор-5-(4-метил-1Н-індол-2-іл)-Nциклогексилбензолсульфонаміду у вигляді твердої речовини (652 мг). РХ/МС: RT=3,11 хв., МС: 403 (М+Н). Стадія 5. Оксалілхлорид (0,21 мл) повільно додають до розчину 2-хлор-5-(4-метил-1Н-індол-2іл)-N-циклогексилбензолсульфонаміду (650 мг) у дихлорметані (16 мл) при кімнатній температурі. Після перемішування протягом ночі додають МеОН (5 мл) і перемішують протягом 15 хв. Суміш концентрують. Залишок очищають флешхроматографією на силікагелі, елююючи 0%-50% EtOAc у гептані для одержання метилового ефіру [2-(4-хлор-3-циклогексилсульфамоїлфеніл)-4метил-1Н-індол-3-іл]оксооцтової кислоти у вигляді жовтої твердої речовини (588 мг). РХ/МС: RT=2,8 хв., МС: 489 (М+Н). Стадія 6. Триетилсилан (0,38 мл) повільно додають до розчину метилового ефіру [2-(4-хлор-3циклогексилсульфамоїлфеніл)-4-метил-1Н-індол3-іл]оксооцтової кислоти (588 мг) у трифтороцтовій кислоті (2 мл) при кімнатній температурі. Після перемішування протягом ночі леткі фракції видаляють під вакуумом. Залишок розчиняють в EtOAc і промивають 1н NaHCO3. Органічну фазу відділяють, сушать над MgSO4 і концентрують. Залишок очищають флеш-хроматографією на силікагелі, елююючи 0%-40% EtOAc у гептані для одержання метилового ефіру [2-(4-хлор-3циклогексилсульфамоїлфеніл)-4-метил-1Н-індол3-іл]оцтової кислоти у вигляді твердої речовини (338 мг). РХ/МС: RT=2,95 хв., МС: 475 (М+Н). Стадія 7. До розчину метилового ефіру [2-(4хлор-3-циклогексилсульфамоїл-феніл)-4-метил1Н-індол-3-іл]оцтової кислоти (330 мг) у МеОН/Н2О (1:1, 7 мл) додають моногідрат гідроксиду літію (58 мг). Реакційну суміш перемішують при температурі 80°С протягом 18 годин. Додають EtOAc (15 мл) і 45 розчин промивають 1н HCl (10 мл). Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання [2-(4-хлор-3циклогексилсульфамоїлфеніл)-4-метил-1Н-індол3-іл]оцтової кислоти у вигляді білої твердої речовини (210 мг). РХ/МС: RT=2,60 хв., МС: 461,12 1 (М+Н); H ЯМР (300 МГц, ДМСО) 1,02-1,28 (м, 5Н), 1,46-1,64 (м, 5Н), 3,07 (м, 1Н), 2,63 (с, 3Н), 3,95 (с, 2Н), 6,79 (д, J=6,9 Гц, 1Н), 7,05 (т, J=8,1 Гц, 1Н), 7,26 (д, J=8,1 Гц, 1Н), 7,80 (м, 2Н), 7,96 (д, J=8,1 Гц, 1Н), 8,21 (с, 1Н), 11,53 (с, 1Н), 12,54 (уш.с, 1Н). IC5О=1,5 нМ. Приклад 8 [7-Хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтова кислота Стадія 1. Ди-трет-бутилдикарбонат (7,92 г) додають до розчину 7-хлоріндолу (5 г) і 4(диметиламіно)піридину (0,4 г) у DCM (165 мл). Реакційну суміш перемішують при кімнатній температурі протягом 18 годин. Реакційну суміш промивають 1н HCl (100 мл) і 1н NHCO3 (100 мл). Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання трет-бутилового ефіру 7-хлоріндол-1-карбонової кислоти у вигляді масла (8,22 г). Стадія 2. До розчину трет-бутилового ефіру 7хлоріндол-1-карбонової кислоти (3 г) у сухому THF (15 мл) додають триізопропілборат (4,11 мл) у присутності азоту. Суміш охолоджують до 0°С у льодяній бані. Додають літійдіізопропіламін (8,94 мл, 2М) протягом години при температурі 0°С. Реакційну суміш перемішують при температурі 0°С протягом 30 хв. Додають 2н HCl (10 мл), щоб погасити реакцію. Одержану суміш екстрагують EtOAc. Залишок очищають флеш-хроматографією на силікагелі, елююючи 10%-50% EtOAc у гептані для одержання 1-(трет-бутоксикарбоніл)-7-хлор-1Ніндол-2-ілборонової кислоти у вигляді твердої речовини (0,86 г). Стадія 3. До розчину 1-(трет-бутоксикарбоніл)7-хлор-1Н-індол-2-ілборонової кислоти (860 мг), 5бром-2-хлор-N-циклогексилбензолсульфонаміду (733 мг) і CsF (632 мг) у діоксан-Н2О (22 мл, 10:1) додають PdCl2(dppf)2 (163 мг) при кімнатній температурі у присутності азоту. Реакційну суміш нагрівають до 80°С і перемішують протягом ночі. Реакційну суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і фільтрують через коротку колонку з силікагелем. Фільтрат концентрують у вакуумі і очищають флеш-хроматографією на силікагелі, елююючи від 10% до 50% EtOAc у гептані для одержання трет-бутилового ефіру 7-хлор-2-(4хлор-3-циклогексилсульфамоїлфеніл)індол-1карбонової кислоти у вигляді твердої речовини (630 мг). 95303 46 Стадія 4. Трифтороцтову кислоту (3 мл) додають до розчину трет-бутилового ефіру 7-хлор-2-(4хлор-3-циклогексилсульфамоїлфеніл)індол-1карбонової кислоти (630 мг) у дихлорметані (7 мл). Реакційну суміш перемішують при кімнатній температурі протягом ночі. Суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають 1н NaHCO3. Органічну фазу відділяють, сушать над MgSO4 і концентрують у вакуумі. Сирий продукт очищають флеш-хроматографією на силікагелі, елююючи 10%-40% EtOAc у гептані для одержання 2-хлор-5-(7-хлор-1Н-індол-2-іл)-Nциклогексилбензолсульфонаміду у вигляді твердої речовини (386 мг). Стадія 5. Оксалілхлорид (0,12 мл) повільно додають до розчину 2-хлор-5-(7-хлор-1Н-індол-2іл)-N-циклогексилбензолсульфонаміду (386 мг) у дихлорметані (9 мл) при кімнатній температурі. Після перемішування протягом 18 годин додають МеОН (3 мл) і перемішують протягом 15 хв. Суміш концентрують. Залишок очищають флешхроматографією на силікагелі, елююючи 5%-45% EtOAc у гептані для одержання метилового ефіру [7-хлор-2-(4-хлор-3-циклогексилсульфамоїлфеніл)-1Н-індол-3-іл]оксооцтової кислоти у вигляді твердої речовини (239 мг). Стадія 6. Триетилсилан (0,15 мл) повільно додають до розчину метилового ефіру [7-хлор-2-(4хл ор-3-циклогексилсульфамоїл феніл)-1Н-індол3-іл]оксооцтової кислоти (239 мг) у трифтороцтовій кислоті (2,4 мл) при кімнатній температурі. Після перемішування протягом ночі леткі фракції видаляють під вакуумом. Залишок розчиняють в EtOAc і промивають 1N NaHCO3. Органічну фазу відділяють, сушать над MgSO4 і концентрують. Залишок очищають флеш-хроматографією на силікагелі, елююючи 10%-50% EtOAc у гептані для одержання метилового ефіру [7-хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти у вигляді твердої речовини (93 мг). РХ/МС: RT=4,5 хв., МС: 495 (М+Н). Стадія 7. До розчину метилового ефіру [7хлор-2-(4-хлор-3-циклогексилсульфамоїлфеніл)1Н-індол-3-іл]оцтової кислоти (93 мг) у МеОН/Н2О (1:1, 4 мл) додають моногідрат гідроксиду літію (16 мг). Реакційну суміш перемішують при температурі 80°С протягом 18 годин. Додають EtOAc (10 мл) і розчин промивають 1н HCl (5 мл). Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання [7-хлор-2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти у вигляді твердої речовини (85 1 мг). РХ/МС: Rt=2,6 хв., МС: 481 (М+Н); Н ЯМР (300 МГц, ДМСО) 1,09-1,35 (м, 5Н), 1,59-1,73 (м, 5Н), 3,19 (м, 1Н), 3,84 (уш.с, 2Н), 7,21 (м, 1Н), 7,38 (м, 1Н), 7,67 (м, 1Н), 7,95 (м, 1Н), 8,02-8,05 (м, 2Н), 8,40 (уш.с, 1Н), 11,9 (уш.с, 1Н). ІС50=3,7 нМ. Приклад 9 2-Хлор-N-циклогексил-5-[3-(2метансульфоніламіно-2-оксоетил)-1Н-індол-2ілібензолсульфонамід 47 Стадія 1. До розчину 1-(трет-бутоксикарбоніл)1Н-індол-2-ілборонової кислоти (10 г), 5-бром-2хлор-N-циклогексилбензолсульфонаміду (6,8 г) і CsF (5,8 г) у діоксан-Н2О (220 мл, 10:1) додають PdCl2(dppf)2 (1,57 г) при кімнатній температурі у присутності азоту. Реакційну суміш нагрівають до 80°С і перемішують протягом 6 годин. Реакційну суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і фільтрують через коротку колонку з силікагелем. Фільтрат концентрують у вакуумі і очищають флеш-хроматографією на силікагелі, елююючи 10%-50% EtOAc у гептані для одержання трет-бутилового ефіру 2-(4-хлор-3циклогексилсульфамоїлфеніл)індол-1-карбонової кислоти у вигляді твердої речовини (8,2 г). Стадія 2. Трифтороцтову кислоту (65 мл) додають до розчину трет-бутилового ефіру 2-(4хлор-3-циклогексилсульфамоїлфеніл)індол-1карбонової кислоти (13 г) у дихлорметані (150 мл). Реакційну суміш перемішують при кімнатній температурі протягом 2 годин. Суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають 1н NaHCO3. Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання 2хлор-5-(1Н-індол-2-іл)-Nциклогексилбензолсульфонаміду у вигляді твердої речовини (9,7 г). РХ/МС: RT = 3,17 хв., МС: 389 (М+Н). Стадія 3. Оксалілхлорид (0,33 мл) повільно додають до розчину 2-хлор-5-(1Н-індол-2-іл)-Nциклогексилбензолсульфонаміду (1 г) у дихлорметані (25 мл) при кімнатній температурі. Після перемішування протягом 18 годин додають МеОН (5 мл) і перемішують протягом 15 хв. Суміш концентрують. Залишок очищають флеш-хроматографією на силікагелі, елююючи 10%-45% EtOAc у гептані для одержання метилового ефіру [2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оксооцтової кислоти у вигляді твердої речовини (1,2 г). Стадія 4. Триетилсилан (0,59 мл) повільно додають до розчину метилового ефіру [2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оксооцтової кислоти (1,2 г) у трифтороцтовій кислоті (12 мл) при кімнатній температурі. Після перемішування протягом ночі леткі фракції видаляють під вакуумом. Залишок розчиняють в EtOAc і промивають 1N NaHCO3. Органічну фазу відділяють, сушать над MgSO4 і концентрують. Залишок очищають флеш-хроматографією на силікагелі, елююючи 10%-50% EtOAc у гептані для одержання метилового ефіру [2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти у вигляді твердої речовини (818 мг). Стадія 5. До розчину метилового ефіру [2-(4хлор-3-циклогексилсульфамоїл-феніл)-1Н-індол-3іл]оцтової кислоти (818 мг) у МеОН/Н2О (1:1, 18 95303 48 мл) додають моногідрат гідроксиду літію (149 мг). Реакційну суміш перемішують при температурі 80°С протягом 18 годин. Додають EtOAc (15 мл) і розчин промивають 1н HCl (10 мл). Органічний шар відділяють, сушать над MgSO4 і концентрують для одержання [2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти у вигляді твердої речовини (740 мг). Стадія 6. До розчину [2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти (185 мг), гідрохлориду N-(3диметиламінопропіл)-N'-етилкарбодііміду (82 мг) і диметиламінопіридину (50 мг) у дихлорметані (4 мл) додають метансульфонамід (41 мг) при температурі 0°С. Реакційну суміш доводять до кімнатної температури і перемішують протягом ночі. Одержаний розчин концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають 1н HCl. Органічну фазу відділяють, сушать над MgSO4 і концентрують у вакуумі. Сирий залишок стирають у порошок з дихлорметаном і фільтрують для одержання 2-хлор-N-циклогексил-5-[3-(2метансульфоніламіно-2-оксоетил)-1Н-індол-2іл]бензолсульфонаміду у вигляді твердої речовини 1 (115 мг). РХ/МС: Rf=2,39 хв., МС: 524 (М+Н); H ЯМР (300 МГц, ДМСО) 1,10-1,28 (м, 5Н), 1,611,64 (м, 5Н), 3,07 (м, 1Н), 3,26 (с, 3Н), 3,88 (с, 2Н), 7,10 (м, 1Н), 7,21 (м, 1Н), 7,44 (м, 1Н), 7,61 (м, 1Н), 7,82 (м, 1Н), 7,98 (м, 2Н), 8,25 (с, 1Н), 11,65 (с, 1Н), 12,12 (с, 1Н). ІС50=2 нМ. Приклад 10 2-Хлор-N-циклогексил-5-[3-(2етансульфоніламіно-2-оксоетил)-1Н-індол-2іл]бензолсульфонамід Стадія 1. До розчину [2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти (200 мг), гідрохлориду N-(3диметиламінопропіл)-N'-етилкарбодііміду (90 мг) і диметиламінопіридину (55 мг) у дихлорметані (4,5 мл) додають етансульфонамід (51 мг) при температурі 0°С. Реакційну суміш доводять до кімнатної температури і перемішують протягом ночі. Одержаний розчин концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають 1н HCl. Органічний шар відділяють, сушать над MgSO4 і концентрують у вакуумі. Сирий залишок стирають у порошок з дихлорметаном і фільтрують для одержання 2-хлор-N -циклогексил-5-[3-(2етансульфоніламіно-2-оксоетил)-1Н-індол-2іл]бензолсульфонаміду у вигляді твердої речовини 1 (174 мг). РХ/МС: RT=2,44 хв., МС: 538 (М+Н); H ЯМР (300 МГц, ДМСО) 1,07-1,33 (м, 8Н), 1,511,69 (м, 5Н), 3,13 (м, 1Н), 3,34 (м, 2Н), 3,94 (с, 2Н), 7,14 (т, J=7,2 Гц, 1Н), 7,26 (т, J=7,2 Гц, 1Н), 7,50 (д, J=7,2 Гц, 1Н), 7,67 (д, J=7,2 Гц, 1Н), 7,87 (д, J=7,2 49 Гц, 1Н), 8,00 (м, 1Н), 8,34 (м, 1Н), 11,70 (с, 1Н), 12,06 (с, 1Н). IC50=2,7 нМ. Приклад 11 2-Хлop-N-циклогексил-5-[3-(2-оксо-2трифторметансульфоніламіноетил)-1Н-індол-2іл]бензолсульфонамід Стадія 1. До розчину [2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти (150 мг), гідрохлориду N-(3диметиламінопротл)-N'-етилкарбодііміду (68 мг) і диметиламінопіридину (40 мг) у дихлорметані (4 мл) додають трифторметансульфонамід (52 мг) при температурі 0°С. Реакційну суміш доводять до кімнатної температури і перемішують протягом ночі. Одержаний розчин концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають 1н HCl. Органічну фазу відділяють, сушать над MgSO4 і концентрують у вакуумі для одержання 2-хлор-Nциклогексил-5-[3-(2-трифторметансульфоніламіно2-оксоетил)-1Н-індол-2-іл]бензолсульфонаміду у вигляді твердої речовини (206 мг). РХ/МС: RT-2,58 1 хв., МС: 576 (М+Н); Н ЯМР (300 МГц, CD3OD) 1,17-1,27 (м, 5Н), 1,55-1,75 (м, 5Н), 3,12 (м, 1Н), 4,01 (с, 2Н), 7,11 (м, 1Н), 7,42 (м, 1Н), 7,50 (м, 1Н), 7,68 (м, 1Н), 7,80 (м, 1Н), 8,3 (м, 1Н). ІС50=14 нМ. Приклад 12 2-[2-(4-Хлор-3-циклогексилсульфамоїлфеніл)1Н-індол-3-іл]-N-(1Н-тетразол-5-іл)ацетамід Стадія 1. До розчину [2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти (200 мг), гідрохлориду N-(3диметиламінопропіл)-N-етилкарбодііміду (90 мг) і диметиламінопіридину (55 мг) у дихлорметані (4,5 мл) додають 1Н-тетразол-5-іламін (48 мг) при температурі 0°С. Реакційну суміш доводять до кімнатної температури і перемішують протягом 2 днів. Одержаний розчин концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають 1н HCl. Органічну фазу відділяють, сушать над MgSO4 і концентрують у вакуумі. Сирий залишок стирають у порошок з дихлорметаном і фільтрують для одержання 2-[2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3-іл]-N(1Н-тетразол-5-іл)ацетаміду у вигляді твердої речовини (50 мг). РХ/МС: RT=2,26 хв., МС: 514 (М+Н); 1 H ЯМР (300 МГц, DMSO) 1,14-1,3 (м, 5Н), 1,51 95303 50 1,65 (м, 5Н), 3,1 (м, 1Н), 4,1 (с, 2Н), 7,11 (м, 1Н), 7,23 (м, 1Н), 7,48 (м, 1Н), 7,7 (м, 1Н), 7,84 (м, 1Н), 8,06 (м, 2Н), 8,34 (с, 1Н), 11,69 (уш.с, 1Н), 12,46 (уш.с, 1Н). ІС50=15 нМ. Приклад 13 [2-(3-Циклогексилсульфамоїл-4-етилфеніл)1Н-індол-3-іл]оцтова кислота Стадія 1. 1-Бром-4-етилбензол (3 г) розчиняють у 30 мл DCM і охолоджують до 0°С у льодяній бані. Хлорсульфонову кислоту (11,3 г) додають по краплях протягом 20 хв. і розчин перемішують при температурі 0°С протягом 4 годин. Реакційну суміш обережно виливають на лід і доводять до кімнатної температури. Суміш переносять у ділильну лійку і розділяють шари. Водний шар промивають додатковим DCM. Органічні шари об'єднують, сушать (MgSO4) і випарюють для одержання 5-бром2-етилбензолсульфонілхлориду (1,78 г) у вигляді масла, яке використовується без подальшого очищення у стадії 2. Стадія 2. Циклогексиламін (0,9 г) і діізопропілетиламін (1,5 г) розчиняють у 20 мл DCM і розчин охолоджують до 0°С. До нього додають порціями 5-бром-2-етилбензолсульфонілхлорид (1,7 г у 20 мл DCM) протягом 5 хв. Суміш перемішують при температурі 0°С протягом 30 хв. і при кімнатній температурі протягом 1 години. Розчинник видаляють при зниженому тиску і до залишку додають 10% водну HCl і DCM. Фази розділяють і водну фазу промивають додатковим DCM. Об'єднані фази DCM сушать (MgSO4), фільтрують і випарюють. Одержану тверду речовину перекристалізовують з DCM/гептану для одержання 5-бром-Nциклогексил-2-етилбензолсульфонаміду (1,36 г). РХ/МС: RT=2,96 хв., МС: 346 (М+Н). Стадія 3. 5-Бром-N-циклогексил-2етилбензолсульфонамід (1,3 г), 1-Вос-індол-2боронову кислоту (1,48 г) і цезію фторид (0,86 г) змішують з 10:1 діоксан:Н2О (44 мл). Розчин дегазують азотом і додають PdCl2(dppf)2 (0,31 г). Суміш нагрівають до 80°С протягом 2,5 години. Реакційну суміш виливають у воду. Додають EtOAc і розділяють фази. Фазу EtOAc концентрують. Додають гептан для одержання осаду, який видаляють фільтрацією. Фільтрат EtOAc пропускають через шар силікагелю, випарюють на силікагелі і очищають на 80 г колонці силікагелю, використовуючи систему очищення ISCO Companion (градієнт EtOAC/гептан), для одержання трет-бутилового ефіру 2-(3-циклогексилсульфамоїл-4етилфеніл)індол-1-карбонової кислоти (1,19 г). РХ/МС: RT=3,54 хв., МС: 483 (М+Н). Стадія 4. Трет-бутиловий ефір 2-(3циклогексилсульфамоїл-4-етилфеніл)індол-1карбонової кислоти (1,18 г) обробляють 10 мл TFA протягом 30 хв. при кімнатній температурі. TFA видаляють при зниженому тиску. Залишок стру 51 шують з EtOAc і 10% водною NaHCO3 і розділяють фази. Органічну фазу промивають додатковим водним розчином NaHCO3, водою і сольовим розчином. Органічний шар сушать (MgSO4), фільтрують, випарюють на силікагелі і очищають на 40 г колонці силікагелю, використовуючи систему очищення ISCO Companion (градієнт EtOAC/гептан), для одержання N-циклогексил-2-етил-5-(1Н-індол2-іл)бензолсульфонаміду (0,65 г). РХ/МС: Rt=3,07 хв., МС: 383 (М+Н). Стадія 5. N-Циклогексил-2-етил-5-(1Н-індол-2іл)бензолсульфонамід (0,64 г) суспендують у 30 мл діетилового ефіру. Оксалілхлорид (0,32 г) додають по краплях при кімнатній температурі і суміш перемішують протягом 6 годин. Додають метанол (2 мл), розчин перемішують протягом 10 хв. і видаляють при зниженому тиску. Сирий матеріал очищають на 80 г колонці силікагелю, використовуючи систему очищення ISCO Companion (градієнт EtOAc/гептан), для одержання метилового ефіру [2-(3-циклогексилсульфамоїл-4-етилфеніл)1Н-індол-3-іл]оксооцтової кислоти (0,66 г). РХ/МС: RT=2,75 хв., МС: 469 (М+Н). Стадія 6. Метиловий ефір [2-(3циклогексилсульфамоїл-4-етилфеніл)-1Н-індол-3іл]оксооцтової кислоти (0,63 г) розчиняють у 10 мл TFA. Триетилсилан (0,31 г) додають по краплях при кімнатній температурі і розчин перемішують протягом 18 годин. Реакційну суміш концентрують при зниженому тиску. До залишку додають EtOAc і насичену NaHCO3 і розділяють фази. Фазу EtOAc випарюють на силікагелі і очищають на 40 г колонці силікагелю, використовуючи систему очищення ISCO Companion (градієнт EtOAc/гептану), для одержання метилового ефіру [2-(3циклогексилсульфамоїл-4-етилфеніл)-1Н-індол-3іл]оцтової кислоти (0,53 г). РХ/МС: RT=2,95 хв, МС: 1 455 (М+Н); H ЯМР (300 МГц, CDCl3) 1,05-1,29 (м, 5Н), 1,37 (т, J=7,5 Гц, 3Н), 1,50-1,79 (м, 5Н), 3,14 (кв, J=7,5 Гц, 2Н), 3,22 (м, 1Н), 3,73 (с, 3Н), 3,84 (с, 2Н), 4,55 (д, J=7,9 Гц, 1Н), 7,16-7,28 (м, 2Н), 7,41 (д, J=8,1 Гц, 1Н), 7,51 (д, J=8,1 Гц, 1Н), 7,69 (д, J=7,7 Гц, 1Н), 7,83 (дд, J=7,9, 1,8 Гц, 1Н), 8,28 (д, J=1,9 Гц, 1Н), 8,32 (с, 1Н). Стадія 7. Метиловий ефір [2-(3циклогексилсульфамоїл-4-етилфеніл)-1Н-індол-3іл]оцтової кислоти (0,33 г) розчиняють у 6 мл 3:3:1 MeOH:THF:H2O. Додають моногідрат гідроксиду літію (2 екв.) і нагрівають розчин до 80°С протягом ночі. Розчинник випарюють при зниженому тиску. Додають EtOAc і 10% водної HCl і розділяють фази. Фазу EtOAc промивають додатково 10% водною HCl, водою і сольовим розчином. Органічний шар сушать (MgSO4), фільтрують, випарюють і залишок перекристалізовують з DCM/гептану для одержання [2-(3-циклогексилсульфамоїл-4етилфеніл)-1Н-індол-3-іл]оцтової кислоти у вигляді твердої речовини (193 мг). РХ/МС: RT=2,6 хв, МС: 1 441 (М+Н); H ЯМР (300 МГц, ДМСО-D6) 1,0-1,24 (м, 5Н), 1,31 (т, J=7,5 Гц, 3Н), 1,45-1,62 (м, 5Н), 3,05 (м, 1Н), 3,08 (кв, J=7,4 Гц, 2Н), 3,75 (с, 2Н), 7,07 (т, J=7,8 Гц, 1Н), 7,18 (т, J=7,7 Гц, 1Н), 7,42 (д, J=8 Гц, 1Н), 7,59 (м, 2Н), 7,76 (д, J=7,9 Гц, 1Н), 7,87 (д, J=7,9 Гц, 1Н), 8,18 (с, 1Н), 11,46 (с, 1Н), 12,37 (с, 1Н). IC50=0,5 нМ. 95303 52 Приклад 14 2-[2-(4-Хлор-3-циклогексилсульфамоїлфеніл)1Н-індол-3-іл]пропіонова кислота Стадія 1. До розчину 1-(третбутоксикарбоніл)індол-2-ілборонової кислоти (5,5 г), 5-бром-2-хлор-Nциклогексилбензолсульфонаміду (5 г) і CsF (4,3 г) у діоксан-Н2О (143 мл, 10:1) додають PdCl2(dppf)2 (1,16 г) при кімнатній температурі у присутності азоту. Реакційну суміш нагрівають до 80°С і перемішують протягом 18 годин. Реакційну суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і фільтрують через коротку колонку з силікагелем. Фільтрат концентрують у вакуумі і очищають флеш-хроматографією на силікагелі, елююючи 5%-30% EtOAc у гептані для одержання третбутилового ефіру 2-(4-хлор-3-циклогексилсульфамоїлфеніл)індол-1-карбонової кислоти у вигляді твердої речовини (6,2 г). РХ/МС: RT=5,03 хв, МС: 511 (M+Na). Стадія 2. Трифтороцтову кислоту (10 мл) додають до розчину трет-бутилового ефіру 2-(4хлор-3-циклогексилсульфамоїлфеніл)індол-1карбонової кислоти (6,2 мг) у дихлорметані (20 мл). Реакційну суміш перемішують при кімнатній температурі протягом ночі. Суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають 1N NaHCO3. Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання 2хлор-N-циклогексил-5-(1Н-індол-2іл)бензолсульфонаміду у вигляді твердої речовини (5,3 г). Стадія 3. Оксалілхлорид (1,59 мл) повільно додають до розчину 2-хлор-N-циклогексил-5-(1Ніндол-2-іл)бензолсульфонаміду (4,8 мг) у дихлорметані (120 мл) при кімнатній температурі. Після перемішування протягом 3 годин додають МеОН (10 мл) і перемішують протягом 15 хв. Суміш концентрують. Залишок очищають флешхроматографією на силікагелі, елююючи 5%-50% EtOAc у гептані для одержання метилового ефіру [2-(4-хлор-3-циклогексилсульфамоїл-феніл)-1Ніндол-3-іл]оксооцтової кислоти у вигляді твердої речовини (2,5 г). Стадія 4. Триетилсилан (1,7 мл) повільно додають до розчину метилового ефіру [2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оксооцтової кислоти (2,5 г) у трифтороцтовій кислоті (25 мл) при кімнатній температурі. Після перемішування протягом ночі леткі фракції видаляють під вакуумом. Залишок розчиняють в EtOAc і промивають 1н NaHCO3. Органічну фазу відділяють, сушать над MgSO4 і концентрують. Залишок очищають флеш-хроматографією на силікагелі, елююючи 10%-50% EtOAc у гептані для одержання метилового ефіру [2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3 53 іл]оцтової кислоти у вигляді твердої речовини (1,84 г). РХ/МС: RT=4,14 хв., МС: 461 (М+Н). Стадія 5. Ди-трет-бутилдикарбонат (807 мг) додають до розчину метилового ефіру [2-(4-хлор3-циклогексилсульфамоїлфеніл)-1Н-індол-3іл]оцтової кислоти (775 мг), триетиламіну (0,52 мл) і 4-(диметиламіно)піридину (42 мг) у DCM (17 мл). Реакційну суміш перемішують при кімнатній температурі протягом 2 днів. Реакційну суміш промивають 1н HCl (10 мл) і 1н NHCO3 (10 мл). Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання трет-бутилового ефіру 2-[4-хлор-3-(N-третбутилоксикарбоніл)циклогексилсульфамоїлфеніл]3-метоксикарбонілметиліндол-1-карбонової кислоти (1,03 г). Стадія 6. До розчину трет-бутилового ефіру 2[4-хлор-3-(N-третбутилоксикарбоніл)циклогексилсульфамоїлфеніл]3-метоксикарбонілметил-індол-1-карбонової кислоти (864 мг) у DMF (13 мл) додають порціями NaH (157 мг) при температурі 0°С. Одержану суміш перемішують при температурі 0°С протягом 15 хв. і додають МеІ (0,82 мл) при температурі 0°С. Реакційну суміш доводять до кімнатної температури і перемішують протягом 3 годин. Реакцію гасять, додаючи насичену NH4CI (10 мл). Суміш екстрагують EtOAc (20 мл). Органічну фазу промивають водою (10 мл) 3 рази, відділяють, сушать над MgSO4 і концентрують. Залишок очищають флеш-хроматографією на силікагелі, елююючи 10%-45% EtOAc у гептані для одержання третбутилового ефіру 2-[4-хлор-3-(N-третбутилоксикарбоніл)циклогексилсульфамоїлфеніл]3-(1-метоксикарбоніл-етил)індол-1-карбонової кислоти у вигляді білої твердої речовини (400 мг). РХ/МС: RT=4,3 хв., МС: 675 (М+Н). Стадія 7. Трифтороцтову кислоту (2 мл) додають до розчину трет-бутилового ефіру 2-[4-хлор-3(N-третбутилоксикарбоніл)циклогексилсульфамоїлфеніл]3-(1-метоксикарбонілетил)індол-1-карбонової кислоти (165 мг) у дихлорметані (4 мл). Реакційну суміш перемішують при кімнатній температурі протягом 4 годин. Суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають 1N NaHCO3. Органічну фазу відділяють, сушать над MgSO4 і концентрують. Залишок очищають флешхроматографією на силікагелі, елююючи 10%-50% EtOAc у гептані для одержання метилового ефіру 2-[2-(4-хлор-3-циклогексил-сульфамоїлфеніл)-1Ніндол-3-іл]пропіонової кислоти у вигляді білої твердої речовини (94 мг). РХ/МС: RT=3,2 хв., МС: 475 (М+Н). Стадія 8. До розчину метилового ефіру 2-[2-(4хлор-3-циклогексил-сульфамоїлфеніл)-1Н-індол-3іл]пропіонової кислоти (94 мг) у МеОН/Н2О (1:1, 2 мл) додають моногідрат гідроксиду літію (17 мг). Реакційну суміш перемішують при температурі 80°С протягом 2 годин. Додають EtOAc (10 мл) і промивають розчин 1н HCl (5 мл). Органічну фазу відділяють, сушать над MgSO4 і концентрують для одержання 2-[2-(4-хлор-3циклогексилсульфамоїлфеніл)-1Н-індол-3іл]пропіонової кислоти у вигляді твердої речовини 95303 54 1 (90 мг). РХ/МС: RT=2,88 хв., МС: 461 (М+Н); Н ЯМР (300 МГц, ДМСО) 1,15-1,37 (м, 5Н), 1,511,71 (м, 8Н), 3,13 (м, 1Н), 4,06 (м, 1Н), 7,02 (т, J=7,2 Гц, 1Н), 7,14 (т, J=6,9 Гц, 1Н), 7,38 (д, J=8,1 Гц, 1Н), 7,67 (м, 2Н), 7,84 (д, J=8,1 Гц, 1Н), 8,37 (с, 1Н). IC50=5,3 нМ. Приклад 15 {2-[4-Хлор-3-(3хлорфенілметансульфоніл)феніл]-1Н-індол-3іл)оцтова кислота Стадія 1. Сульфіт натрію (1,7 г) і двохосновний фосфат натрію (0,98 г) розчиняють у 20 мл води і нагрівають до 30°С до повного розчинення. Додають 5-бром-2-хлорбензолсульфонілхлорид (2 г) і реакційну суміш нагрівають до 60°С протягом ночі. Реакційну суміш охолоджують і додають 1бромметил-3-хлорбензол (1,4 г) по краплях у вигляді розчину у 20 мл ацетону. Суміш нагрівають до 60°С протягом 2 годин і доводять до кімнатної температури. Реакційну суміш струшують з EtOAc і водою і розділяють фази. Водну фазу промивають додатковим EtOAc. Об'єднані органічні фази промивають водою і сольовим розчином. Органічний шар сушать (MgSO4), фільтрують і випарюють. Сирий залишок перекристалізовують з EtOAc/гептану для одержання 4-бром-1-хлор-2-(3хлорфенілметансульфоніл)бензолу (1,47 г). 1 РХ/МС: RT=2,8 хв., МС: 379 (M+Na), H ЯМР (300 МГц, CDCl3) 4,37 (с, 2H), 6,70 (уш.с, 1Н), 7,157,35 (м, 6Н), 7,73 (д, J=2 Гц, 1Н). Стадія 2. До розчину 4-бром-1-хлор-2-(3хлорфенілметансульфоніл)бензолу (1 г), 1-(третбутоксикарбоніл)індол-2-ілборонової кислоти (1 г) і цезію фториду (0,6 г) у 22 мл 10:1 діоксан:води додають PdCl2(dppf)2 (0,216 г) при кімнатній температурі у присутності азоту. Реакційну суміш нагрівають до 80°С протягом ночі. Після охолоджування реакційну суміш виливають у воду і екстрагують EtOAc. Органічну фазу концентрують і додають гептан для одержання осаду, який фільтрують. Фільтрат пропускають через шар силікагелю і потім випарюють на силікагелі. Сирий матеріал очищають флеш-хроматографією на силікагелі (EtOAc/гептан) для одержання трет-бутилового ефіру 2-[4-хлор-3-(3хлорфенілметансульфоніл)феніл]індол-1карбонової кислоти (0,84 г). РХ/МС: RT=3,42 хв., МС: 516 (M+Na). Стадія 3. Трифтороцтову кислоту (10 мл) додають до трет-бутилового ефіру 2-[4-хлор-3-(3хлорфенілметансульфоніл)феніл]індол-1карбонової кислоти (0,79 г) і одержаний розчин перемішують протягом 35 хв. при кімнатній температурі. Суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають 10% NaHCO3. Органічний шар сушать (MgSO4), фільтрують, ви 55 парюють на силікагелі і очищають флешхроматографією на силікагелі (EtOAc/гептан) для одержання 2-[4-хлор-3-(3хлорфенілметансульфоніл)феніл]-1Н-індолу (0,49 г). РХ/МС: RT=3 хв., МС: 416 (M+Na). Стадія 4. До суспензії 2-[4-хлор-3-(3хлорфенілметансульфоніл)феніл]-1Н-індолу (0,48 г) у 25 мл Et2O додають по краплях оксалілхлорид (0,22 г) при кімнатній температурі. Через 7 годин додають додаткову кількість оксалілхлориду (0,22 г) і суміш перемішують протягом ночі. Додають по краплях метанол (2 мл) і суміш перемішують протягом 10 хв. Реакційну суміш виливають у воду і екстрагують EtOAc. Органічну фазу промивають водним розчином NaHCO3 і сольовим розчином. Органічний шар сушать (Na2SO4), фільтрують і випарюють на силікагелі. Сирий матеріал очищають флеш-хроматографією на силікагелі (EtOAc/гептан) для одержання метилового ефіру {2-[4-хлор-3-(3-хлорфенілметансульфоніл)феніл]1Н-індол-3-іл}оксооцтової кислоти (0,43 г). РХ/МС: RT=2,73 хв., МС: 502 (M+Na). Стадія 5. Триетилсилан (0,23 г) додають по краплях до розчину метилового ефіру {2-[4-хлор-3(3-хлорфенілметансульфоніл)феніл]-1Н-індол-3іл}оксооцтової кислоти (0,5 г) у 10 мл трифтороцтової кислоти. Після перемішування протягом 5 годин реакційну суміш концентрують при зниженому тиску. Залишок струшують з EtOAc і насиченою NaHCO3. Органічну фазу випарюють на силікагелі і очищають флеш-хроматографією на силікагелі (EtOAc/гептан) для одержання метилового ефіру {2-[4-хлор-3-(3хлорфенілметансульфоніл)феніл]-1Н-індол-3іл}оцтової кислоти (0,34 г). РХ/МС: RT=2,86 хв., МС: 488 (M+Na). Стадія 6. До розчину метилового ефіру {2-[4хлор-3-(3-хлорфенілметансульфоніл)феніл]-1Ніндол-3-іл} оцтової кислоти (0,3 г) у 14 мл 3:3:1 THF:MeOH:H2O додають гідроксид літію (0,077 г). Розчин перемішують при температурі 80°С протягом ночі. Розчинник видаляють при зниженому тиску і до залишку додають 10% водного розчину HCl. Водну фазу двічі екстрагують EtOAc. Об'єднані органічні фази сушать (MgSO4), фільтрують і випарюють для одержання {2-[4-хлор-3-(3хлорфенілметансульфоніл)феніл]-1Н-індол-3-іл} оцтової кислоти (210 мг). РХ/МС: RT=2,43 хв., МС: 1 474,1 (M+Na). H ЯМР (300 МГц, DMSO) 3,61 (с, 2Н), 4,97 (с, 2Н), 7,08 (т, J=7,2 Гц, 1Н), 7,21 (м, 2Н), 7,34-7,44 (м, 4Н), 7,57 (д, J=7,9 Гц, 1Н), 7,95-8,05 (м, 2Н), 8,37 (д, J=2 Гц, 1Н), 11,58 (с, 1Н), 12,43 (с, 1Н). IC50=106нМ. Приклад 16 {2-[4-Хлор-3-(3хлорфенілметансульфоніламіно)феніл]-1Н-індол3-іл}оцтова кислота 95303 56 Стадія 1. До гідрохлоридної солі 5-бром-2хлорфеніламіну (0,81 г) у 20 мл DCM додають Et3N (0,85 г) і розчин охолоджують до 0°С. Порціями додають 3-хлорфенілметансульфонілхлорид (0,75 г) у вигляді розчину у 5 мл DCM. Суміш нагрівають до кімнатної температури і перемішують протягом ночі. Розчинник видаляють при зниженому тиску і залишок повторно розчиняють в EtOAc. EtOAc екстрагують 10% водною HCl, насиченою Na2CO3 і сольовим розчином. Органічний шар сушать (MgSO4), фільтрують, випарюють на силікагелі і очищають флеш-хроматографією на силікагелі (EtOAc/гептан) для одержання N-(5-бром-2хлорфеніл)-С-(3-хлорфеніл)метансульфонаміду (1,56 г). РХ/МС: RT=2,77 хв., МС: 394 (M+Na). Стадія 2. До розчину N-(5-бром-2-хлорфеніл)С-(3-хлорфеніл)метансульфонаміду (0,6 г), 1(трет-бутоксикарбоніл)індол-2-ілборонової кислоти (0,6 г) і цезію фториду (0,35 г) у 10:1 діоксан:воді (11 мл) додають PdCl2(dppf)2 (0,125 г) у присутності азоту. Суміш нагрівають до 80°С протягом 3 годин. Після охолоджування реакційну суміш виливають у воду і екстрагують EtOAc. Органічну фазу концентрують і додають гептан для одержання осаду, який фільтрують. Фільтрат пропускають через шар силікагелю і випарюють на силікагелі. Сирий матеріал очищають флеш-хроматографією на силікагелі (EtOAc/гептан) для одержання третбутилового ефіру 2-[4-хлор-3-(3хлорфенілметансульфоніл-аміно)феніл]індол-1карбонової кислоти (0,73 г). РХ/МС: RT=3,36 хв., МС: 531 (M+Na). Стадія 3. Трифтороцтову кислоту (10 мл) додають до трет-бутилового ефіру 2-[4-хлор-3-(3хлорфенілметансульфоніламіно)феніл]індол-1карбонової кислоти (0,70 г) і одержаний розчин перемішують при кімнатній температурі протягом 1 години. Суміш концентрують у вакуумі. Залишок розчиняють в EtOAc і промивають 10% NaHCO3. Органічну фазу сушать (MgSO4), випарюють на силікагелі і очищають флеш-хроматографією на силікагелі (EtOAc/гептан) для одержання N-[2хлор-5-(1Н-індол-2-іл)феніл]-С-(3хлорфеніл)метансульфонаміду (0,56 г). РХ/МС: RT=2,93 хв., МС: 431 (M+Na). Стадія 4. До суспензії N-[2-хлор-5-(1Н-індол-2іл)феніл]-С-(3-хлорфеніл)метансульфонаміду (0,51 г) у 30 мл DCM додають по краплях оксалілхлорид (0,23 г) при кімнатній температурі. Після перемішування протягом 2 годин додають по краплях метанол (2 мл) і суміш перемішують протягом 10 хв. Реакційну суміш потім випарюють на силікагелі і очищають флеш-хроматографією на силікагелі (EtOAc/гептан) для одержання метилового ефіру {2-[4-хлор-3-(3хлорфенілметансульфоніламіно)феніл]-1Н-індол 57 3-іл}оксооцтової кислоти (0,46 г). РХ/МС: RT=2,67 хв., МС: 517 (M+Na). Стадія 5. Триетилсилан (0,19 г) додають по краплях до розчину метилового ефіру {2-[4-хлор-3(3-хлорфенілметансульфоніламіно)феніл]-1Ніндол-3-іл}оксо-оцтової кислоти (0,42 г) у 10 мл трифтороцтової кислоти. Суміш перемішують протягом 6 годин. Додають додаткову кількість триетилсилану (0,1 г) і розчин перемішують протягом ночі при кімнатній температурі. Суміш потім концентрують при зниженому тиску і залишок струшують з EtOAc і насиченою NaHCO3. Органічну фазу випарюють на силікагелі і очищають флешхроматографією на силікагелі (EtOAc/гептан) для одержання метилового ефіру {2-[4-хлор-3-(3хлорфенілметансульфоніламіно)феніл]-1Н-індол3-іл}оцтової кислоти (0,3 г). РХ/МС: RT=2,86 хв., МС: 503 (M+Na). Стадія 6. До розчину метилового ефіру {2-[4хлор-3-(3-хлорфенілметан-сульфоніламіно)феніл]1Н-індол-3-іл}оцтової кислоти (0,24 г) у 3:3:1 THF:MeOH:H2O (14 мл) додають гідроксид літію (0,041 г). Розчин перемішують при температурі 80°С протягом ночі. Додають додатково 2 еквіваленти гідроксиду літію і продовжують нагрівання протягом 6 годин до завершення реакції. Розчинник видаляють при зниженому тиску і до залишку додають 10% водного розчину HCl. Суміш двічі екстрагують EtOAc. Об'єднані органічні фази сушать (MgSO4), фільтрують, випарюють на силікагелі і очищають флеш-хроматографією на силікагелі (EtOAc/гептан) для одержання {2-[4-хлор-3-(3хлорфенілметан-сульфоніламіно)феніл]-1Н-індол3-іл}оцтової кислоти (186 мг). РХ/МС: RT=2,53 хв., 1 МС: 489 (M+Na). H ЯМР (300 МГц, DMSO) 3,78 (с, 2Н), 4,67 (с, 2Н), 7,07 (т, J=7,2 Гц, 1Н), 7,19 (т, J=7,3 Гц, 1Н), 7,40-7,46 (м, 4H), 7,51 (с, 1Н), 7,57 (м, 2Н), 7,70 (д, J=8,2 Гц, 1Н), 7,83 (с, 1Н), 9,75 (с, 1Н), 11,44 (с, 1Н), 12,44 (с, 1Н). IC50=12 нМ. Фармакологічний аналіз Інгібіторну дію сполук, що складають предмет даного винаходу, оцінюють за допомогою функціонального аналізу на простагландин D людини. Застосовується аналіз цАМФ з використанням лінії клітин людини LS174T, що експресують ендогенний рецептор простагландину D. Протокол аналогічний описаному раніше (Wright DH, FordHutchinson AW, Chadee K, Metters KM, The human prostanoid DP receptor stimulates mucin secretion in LS174T cells, Br J Pharmacol. 131(8): 1537-45 (2000)). Протокол SPA-аналізу цАМФ у клітинах LS174T людини Матеріали - PGD2 (Cayman Chemical, кат. № 12010) - ІВМХ (Sigma кат. № 5879) - Система прямого скринінгового SPA-аналізу цАМФ (код Amersham RPA 559) - 96-ямкові планшети для клітинних культур (Wallac кат. № 1450-516) - Сцинтиляційний лічильник Wallac 1450 Microplate Trilux (PerkinElmer) - Пристосування для заклеювання планшетів - Центрифугові пробірки Eppendorf 95303 58 - Фізіологічний розчин у фосфатному буфері (PBS) Дульбекко (Invitrogen кат. № 14040-133) - Дистильована вода - Струшувач - Магнітна мішалка і набір якорів Підготовка реактивів: Перед розбавленням всі реагенти повинні бути доведені до кімнатної температури. Аналітичний буфер 1Х Переносять вміст пляшки у 500-мл мензурку багаторазовим промиванням дистильованою водою. Доводять повний об'єм до 500 мл, додавши дистильовану воду, і ретельно перемішують. Лізуючий реагент 1 і 2 Розчиняють кожний з лізуючих реагентів 1 і 2 у 200 мл аналітичного буфера, відповідно. Залишають на 20 хвилин при кімнатній температурі для розчинення. Гранули антитіл кролика для SPA-аналізу Додають у пляшку 30 мл лізуючого буфера 2. Обережно струшують пляшку протягом 5 хвилин. Імунна сироватка Додають 15 мл лізуючого буфера 2 у кожну посудину і обережно перемішують вміст до повного розчинення. 125 Маркер (І -цАМФ) Додають 14 мл лізуючого буфера 2 у кожну посудину і обережно перемішують вміст до повного розчинення. Підготовка імунореактиву 1. Додають рівні об'єми маркера, імунної сироватки і SPA-реагенту антитіл кролика у посудину, слідкуючи за тим, щоб був приготований достатній об'єм для потрібної кількості ямок (150 мкл на ямку). 2. Ретельно перемішують. 3. Обов'язково готують свіжий розчин імунореактиву перед кожним аналізом і не використовують його повторно. Стандарт 1. Додають 1 мл лізуючого буфера 1 і обережно перемішують вміст до повного розчинення. 2. Кінцевий розчин містить цАМФ у концентрації 512 пмоль/мл. 3. Мітять 7 поліпропіленових або полістирольних пробірок 0,2 пмоль, 0,4 пмоль, 0,8 пмоль, 1,6 пмоль, 3,2 пмоль, 6,4 пмоль і 12,8 пмоль. 4. Піпеткою додають 500 мкл лізуючого буфера 1 у всі пробірки. 5. У пробірку 12,8 пмоль додають піпеткою 500 мкл вихідного розчину стандарту (512 пмоль/мл) і ретельно перемішують. Переносять 500 мкл з пробірки 12,8 пмоль у пробірку 6,4 пмоль і ретельно перемішують. Повторюють процедуру розбавлення вдвічі з іншими пробірками. 6. Порції по 50 мкл у двох екземплярах з кожного послідовного розбавлення і вихідний розчин стандарту дозволяють одержати 8 рівнів стандартів цАМФ у діапазоні від 0,2 до 25,6 пмоль. Буфер розбавлення сполуки Додають 50 мкл 1 мМ ІВМХ у 100 мл PBS для одержання кінцевої концентрації 100 мкМ і обробляють ультразвуком при 30°С протягом 20 хвилин. Підготовка PGD2 59 95303 Розчиняють 1 мг PGD2 (FW, 352.5) у 284 мкл DMSO для одержання вихідного розчину 10 мМ і зберігають його при 20°С. Перед кожним аналізом обов'язково готують свіжий розчин. Додають 3 мкл 10 мМ вихідного розчину у 20 мл ДМСО, ретельно перемішують і переносять 10 мл у 40 мл PBS. Розбавлення сполуки А В С D Ε F G Η 1 1 2 3 4 5 6 7 контрольний 2 3 4 Переносять 45 мкл DMSO у планшет, крім колонки 7, куди додають 28 мкл ДМСО. Ретельно відміряють піпеткою 12 мкл вмісту колонки 1 і переносять їх у колонку 7. Виконують послідовне розбавлення 1:10 від колонки 1 до колонки 6 і від колонки 7 до колонки 11, переносячи від 5 мкл до 45 мкл ДМСО для одержання наведених нижче концентрацій: Перший планшет Колонка 12 Колонка 11 Колонка 10 Колонка 9 Колонка 8 Колонка 7 Колонка 6 Колонка 5 Колонка 4 Колонка 3 Колонка 2 Колонка 1 Кінцева концентрація 0 0,03 мкМ 0,3 мкМ 3 мкМ 0,03 мМ 0,3 мМ 0,01 мкМ 0,1 мкМ 1 мкМ 0,01 мМ 0,1 мМ 1 мМ Додають у новий 96-ямковий планшет 247,5 мкл буфера розбавлення сполуки. Переносять 2,5 мкл послідовно розбавлених сполук з приготованого вище планшету у новий (розбавлення 1:100) наступним чином: Перший планшет Колонка 12 Колонка 6 Колонка 11 Колонка 5 Колонка 10 Колонка 4 Колонка 9 Колонка 3 Колонка 8 Колонка 2 Колонка 7 Колонка 1 Другий планшет Колонка 1 Колонка 2 Колонка 3 Колонка 4 Колонка 5 Колонка 6 Колонка 7 Колонка 8 Колонка 9 Колонка 10 Колонка 11 Колонка 12 Кінцева концентрація 0 0,1 нМ 0,3 нМ 1 нМ 3 нМ 0,01 мкМ 0,03 мкМ 0,1 мкМ 0,3 мкМ 1 мкМ 3 мкМ 10 мкМ 60 Розбавлення сполуки проводять у Biomex 2000 (Beckman) за допомогою методу 11 точок 1_цАМФ DP. 5 мкл кожної сполуки з 10 мМ планшетів стандартних розчинів сполук переносять в ямки 96ямкового планшету, як зазначено нижче. 5 6 7 8 9 10 11 12 Вирощування клітин 1. LSI74 Τ завжди вирощують у MEM (ATCC кат. № 30-2003), 10% FBS (АТСС кат. № 30-2020) і додаткових 2 мМ L-глутаміну, при 37°С і 5% СО2. 2. Нагрівають 0,05% трипсину і версину (Invitrogen кат. № 25300-054) при 37°С на водяній бані. 3. Видаляють середовище для росту з клітин. Клітини у колбі Τ165 двічі промивають 4 мл трипсину, а потім інкубують при 37°С і 5% СО2 протягом 3 хвилин. 4. Додають 10 мл середовища і піпеткою ретельно відділяють і перераховують клітини. 5 5. Доводять щільність клітин до 2,25x10 клітин на мл і висівають 200 мкл клітин на ямку (45000 клітин на ямку) у 96-ямковому планшеті за 1 день до аналізу. Порядок проведення аналізу День 1 Висівають 45000 клітин на ямку у 200 мкл середовища у 96-ямкових планшетах. Інкубують планшети з клітинами при 37°С, 5% СО2 і 95% вологості протягом ночі. День 2 1. Виконують розбавлення сполук. 2. Готують аналітичний буфер, лізуючі буфери 1 і 2, PGD2 і стандарт. 3. Видаляють середовище з клітин аспіратором і додають 100 мкл розчину сполуки, дотримуючись протоколу цАМФ DP Zymark ScicloneALH/FD. 4. Інкубують клітини при 37°С, 5% СО2 і 95% вологості протягом 15 хвилин. 5. Додають 5 мкл 300 нМ PGD2 (20x15 нМ кінцевої концентрації) у кожну ямку відповідно до протоколу Zymark цАМФ DP PGD2 та інкубують клітини при 37°C, 5% CO2 і 95% вологості ще 15 хвилин. 6. Видаляють середовище з клітин аспіратором і додають 50 мкл лізуючого буфера 1 відповідно до протоколу Zymark цАМФ DP лізису та інкубують при кімнатній температурі зі струшуванням протягом 30 хвилин. 7. Додають у всі ямки 150 мкл імунореагенту (повний об'єм 200 мкл на ямку).
ДивитисяДодаткова інформація
Назва патенту англійською2-phenyl-indoles as prostaglandin d2 receptor antagonists
Автори англійськоюYang Zhaoxia, Reiling Stephan, Nieduzak Thaddeus R., Mathew Rose M., Jackson Sharon, Harris Keith J.
Назва патенту російською2-фенилиндолы как антагонисты рецепторов простагландина d2
Автори російськоюЯн Чжаося, Райлинг Штефан, Недузак Таддеуш Р., Метью Роуз М., Джексон Шерон, Харрис Кит Дж.
МПК / Мітки
МПК: A61K 31/404, C07D 209/18, A61P 11/06, A61P 27/14, A61P 37/08, A61P 37/06
Мітки: 2-феніліндоли, простагландину, антагоністи, рецепторів
Код посилання
<a href="https://ua.patents.su/31-95303-2-fenilindoli-yak-antagonisti-receptoriv-prostaglandinu-d2.html" target="_blank" rel="follow" title="База патентів України">2-феніліндоли як антагоністи рецепторів простагландину d2</a>
2,6-заміщені-4-монозаміщені аміно-піримідини як антагоністи рецептора простагландину d2

Номер патенту: 88485
Опубліковано: 26.10.2009
Автори: Стефані Девід, Хант Хейзел Дж., Цао Бін, Гіллеспі Тімоті А., Гарднер Чарльз Дж., Боффі Рей, Дешо Ельза А., Агіар Жуасі К., Лім Сунгтаєк, Харріс Кейт Джон
МПК: C07D 403/10, C07D 409/12, C07D 239/46, C07D 239/48, C07D 409/04, C07D 401/04, C07D 403/04, C07D 413/10, C07D 413/04, C07D 405/04, C07D 413/12, C07D 403/12, C07D 401/12, C07D 401/14, C07D 413/14
Мітки: 2,6-заміщені-4-монозаміщені, аміно-піримідини, рецептора, антагоністи, простагландину
Формула / Реферат:
1. Сполука формули (І), (I)де(А) Су1 - циклоалкіл, гетероцикліл, циклоалкеніл, гетероцикленіл, гетероарил, арил або поліциклічний алкарил, кожний з яких необов’язково має від однієї до трьох однакових або різних замісних груп Су1, якими є:ацил, ціано, галоген, нітро, карбокси, гідрокси, алкілтіо, алкілсульфоніл, алкілсульфініл, циклоалкіл,...
Похідні тіазолідинкарбоксаміду як модулятори рецепторів простагландину f

Номер патенту: 78021
Опубліковано: 15.02.2007
Автори: Куаттропані Анна, Помель Венсан, Гамелен Естель, Жоран-Лебрен Катрін, Паж Патрік, Томас Расселл Дж., Шварц Маттіас
МПК: A61P 43/00, A61K 31/55, C07D 417/12, A61K 31/4439, A61K 31/5377, C07D 417/14, A61K 31/426, A61P 15/06, A61K 31/427, A61P 15/00, C07D 277/06, A61K 31/454
Мітки: похідні, рецепторів, модулятори, простагландину, тіазолідинкарбоксаміду
Формула / Реферат:
1. Застосування сполуки за формулою II II,а також її геометричних ізомерів, оптично активних форм, наприклад, енантіомерів, діастереомерів, та рацемічних форм, а також її фармацевтично прийнятних солей та фармацевтично активних похідних, деG вибраний з групи, до якої входять С1-С6-алкіларил, C1-C6-алкілгетероарил, С1-С6-алкілциклоалкіл,...
Азабіциклічні, азатрициклічні і азаспіроциклічні похідні аміноциклогексану як антагоністи рецепторів nmda, 5ht і нейронних нікотинових рецепторів

Номер патенту: 77778
Опубліковано: 15.01.2007
Автори: Гольд Маркус, Парсонс Крістофер Грахам Рафаель, Каусс Валерьянс, Даниш Войцех, Хенріх Маркус, Калвіньш Іварс, Ванейєвс Максімс, Йіргенсонс Айгарс
МПК: A61P 9/10, C07D 471/08, A61P 1/04, A61P 25/24, A61P 25/02, A61P 31/18, A61K 31/437, A61P 25/08, C07D 209/02, A61P 1/08, A61P 21/02, A61P 21/00, C07D 487/08, A61P 25/22, A61P 25/14, A61P 25/36, A61P 33/06, A61P 25/04, A61P 31/14, A61P 25/28, A61P 27/06, A61P 25/16, C07D 221/22, A61K 31/438, A61P 31/12, C07D 221/20, A61P 25/34, A61P 25/06, A61P 25/30, C07D 209/54, A61P 25/32, A61K 31/403, A61P 43/00, A61P 27/16, A61P 25/18, A61P 37/02, A61P 9/00, A61P 25/00, A61K 31/439
Мітки: нікотинових, азабіциклічні, nmda, нейронних, азатрициклічні, антагоністи, рецепторів, азаспіроциклічні, похідні, аміноциклогексану
Формула / Реферат:
1. Сполуки формули (1),де R і R1-R5, кожний незалежно, вибраний з С1-6-алкільних груп, С2-6-алкенільних груп, С2-6-алкінільних груп, С6-12-арил-С1-4-алкільних груп, необов'язково заміщених С6-12-арильних груп і у випадку R і R2-R5 - з атомів водню, за умови, що щонайменше один з R2 і R3 і щонайменше один з R4 і R5 не є воднем, або R і R1 разом являють собою...
Похідні триазолу як антагоністи рецепторів тахікінінів

Номер патенту: 79113
Опубліковано: 25.05.2007
Автори: Гардінієр Кевін Мет'ю, Джангхейм Луіс Ніколаус, Гонг Жан Ерік, Робертсон Майкл Алан, Мехл Брайан Стефан, Амегадзі Алберт Кудзові, Савін Кеннет Аллен, Ремік Дейвід Майкл, Хембре Ерік Джеймз
МПК: C07D 403/04, C07D 487/04, A61P 11/06, A61P 43/00, A61P 25/24, A61K 31/5377, C07D 417/14, A61P 25/00, A61P 17/00, A61K 31/4196, A61K 31/501, A61P 1/04, C07D 249/06, A61K 31/4192, A61K 31/541, A61K 31/506, C07D 403/14, A61K 31/497, A61P 25/04, A61K 31/5025, A61K 31/437, A61K 31/444, A61P 1/08, C07D 401/04, A61P 25/22, A61K 31/4439, C07D 401/14, C07D 249/08, C07D 413/04, C07D 498/04, A61P 25/18, A61K 31/4985, C07D 413/14, A61K 31/422
Мітки: тахікінінів, похідні, антагоністи, триазолу, рецепторів
Формула / Реферат:
1. Сполука Формули І:,де:D - С1-С3-алкандіїл;R1 - феніл,факультативно заміщений замісниками в кількості від одного до трьох, незалежно один від одного вибраними з групи, до якої входять галоген, С1-С4-алкіл, С1-С4-алкокси-, ціаногрупа, дифторметил, трифторметил та трифторметоксигрупа;R4 - радикал, вибраний з групи, до якої...
Похідні імідазолу як антагоністи глутаматних рецепторів

Номер патенту: 80888
Опубліковано: 12.11.2007
Автори: Колчевскі Сабіне, Віера Ерік, Буеттелманн Бернд, Портер Річард Х'ю Філліп, Сессареллі Сімона Маріа, Ешке Георг
МПК: A61K 31/444, C07D 401/14, A61K 31/4439, A61P 25/28, C07D 401/06
Мітки: похідні, глутаматних, антагоністи, рецепторів, імідазолу
Формула / Реферат:
1. Сполука загальної формули, (I)де R1 означає галоген або ціано;R2 означає С1-С6-алкіл;R3 означає феніл або 5- або 6-членний гетероарил, який необов'язково заміщений одним, двома або трьома замісниками, вибраними з групи, що містить галоген, С1-С6-алкіл, С3-С6-циклоалкіл, С1-С6-алкілгалоген, ціано, С1-С6-алкокси, NR'R",...