Карбоксамідні похідні піперидину, композиція на їх основі та спосіб лікування тромбоцитарних розладів
Номер патенту: 70285
Опубліковано: 15.10.2004
Автори: Хоекстре Вільям Дж., Костанзо Майкл Дж., Марянофф Брюсс Е.






Формула / Реферат
1. Карбоксамідні похідні піперидину загальної формули (I):
,
де R10 - це Н або C(O)N(R1)YZ,
де R1 – це Н; Y - це (СН2)р, (CH2)qCH(R3) або CH(R3)(CH2)q ;
де R3 - це арил, аралкіл або гетероарил; q = 1, 2 або 3; р = 2 або 3; Z - це СО2Н, СО2алкіл або 5-тетразол;
Х - це С(О);
М - це (СН2)m або піперидин-1-іл; де m = 2; n = 2;
R5 - це Н;
А вибирають з піперидин-2-ілу, піперидин-3-ілу, піперидин-4-ілу або
,
де R9 – це Н, алкіл, СН(NH), CMe(NH) або ацил;
причому, якщо R10 в положенні 3 - C(O)N(R1)YZ, де R1 - H; Y - (CH2)2; Z - CO2H, то М не є (СН2)2; А не є
,
де R9 – Н;
їх енантіомери або фармацевтично прийнятні солі.
2. Сполука за п. 1, вибрана із:
N-3-(4-піперидинпропіоніл)ніпекотил(3-аміно-3-феніл)пропіонової кислоти,
N-3-(4-піперидинпропіоніл)ізоніпекотил[(3-аміно-3-(4-карбоксифеніл)]пропіонової кислоти,
N-3-(4-піперидинпропіоніл)ніпекотил-5Н-(2-аміноетил)тетразолу,
N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил[(S)-3-аміно-3-(3,4-метилендіоксифеніл)]пропіонової кислоти,
N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил[(S)-3-аміно-3-(3-хінолініл)]пропіонової кислоти,
N-3-(4-піперидинпропіоніл)ніпекотил-[(S)-3-аміно-3-(3,4-метилендіоксифеніл)]пропіонової кислоти,
N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил[(S)-3-аміно-3-(піридил)]пропіонової кислоти,
N-[(4,4’-біпіперидин-1-іл)карбоніл]-R-(-)-ніпекотил[(S)-3-аміно-3-(3-піридил)]пропіонової кислоти,
N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил[(S)-3-аміно-3-(6-метил-3-піридил)]пропіонової кислоти,
N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил[(S)-3-аміно-3-(5-бром-3-піридил)]пропіонової кислоти та
N-3-(4-формамідинопіперидинопропіоніл)-R-(-)-ніпекотил[(S)-3-аміно-3-(3-піридил)]пропіонової кислоти.
3. Композиція для лікування тромбоцитарних розладів, що містить сполуку за п. 1 в кількості, ефективній для лікування таких розладів, в комбінації з фармацевтично прийнятним носієм.
4. Спосіб лікування тромбоцитарних розладів, що включає призначення пацієнту ефективної для лікування таких розладів кількості сполуки за п. 1.
5. Спосіб за п. 4, де кількість сполуки за п. 1 складає 0,1 - 300 мг/кг/день.
Текст
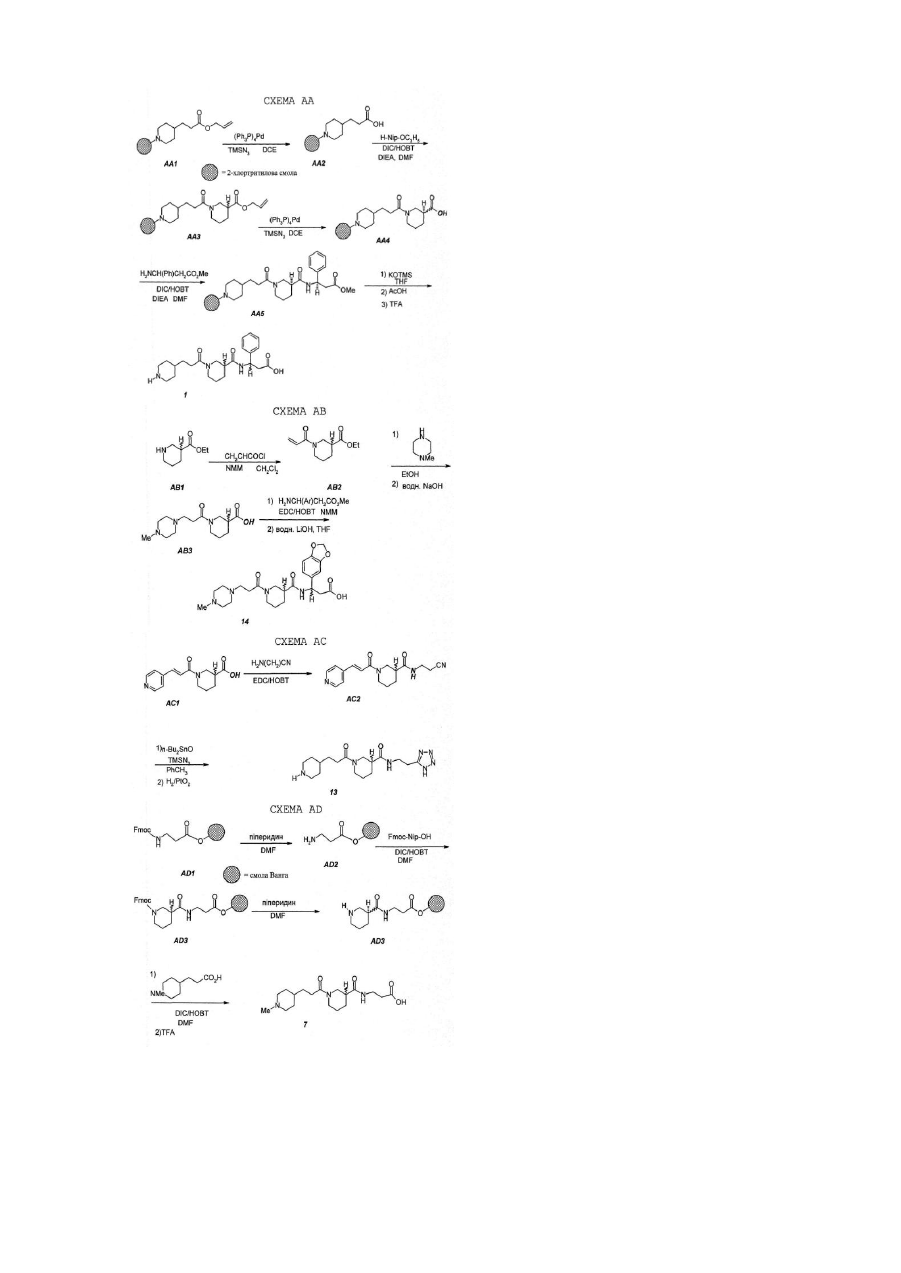
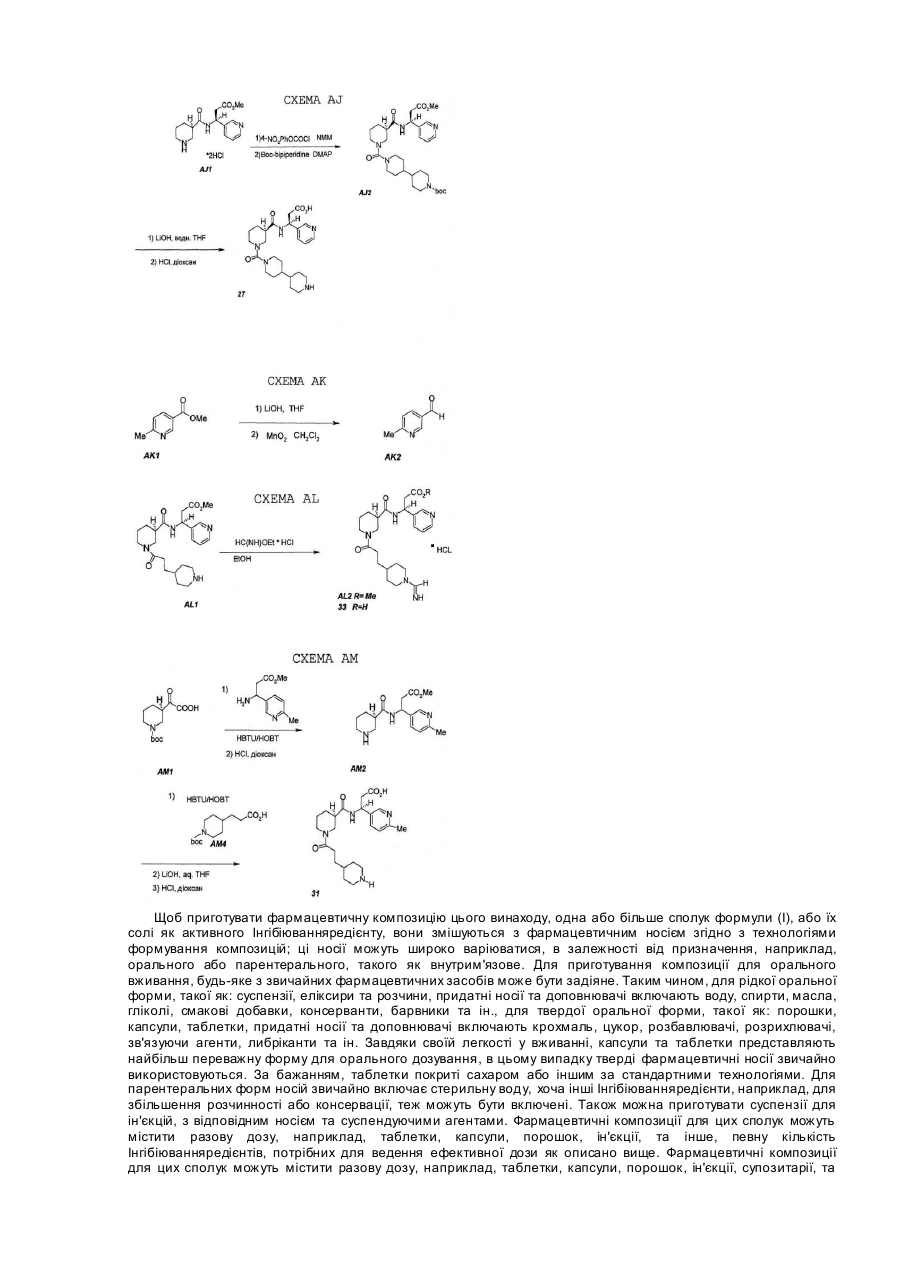
Агрегація тромбоцитів складає початкову стадію механізму зупинення кровотечі, спричиненої ураженням судини. Але, патологічний ріст цього нормального процесу кровоспинення може привести до утворення тромбів. Кінцева ступінь в агрегації тромбоцитів - це зв'язування фібриногену з активованим глікопротеїном тромбоциту ІІв/ІІІа (GP ІІв/ІІІа). Агенти, що заважають зв'язуванню фібриногену з GP ІІв/ІІІа, сповільнюють агрегацію тромбоцитів. Внаслідок цього, такі агенти можуть бути корисними для лікування хвороб, зв’язаних з тромбоцитами, таких як артеріальні та венозні тромбози, гострий інфаркт міокарда, нестійка ангіна, закупорка судин, разом з тромболітичню терапією та ангіопластикою, запалення та ряд хвороб, зв’язаних з закупоркою судин. Рецептор фібриногену (GP ІІв/ІІІа) активується такими стимуляторами, як АДФ, колаген, та за зв'язування з тромбіном відповідають дві різні білкових ділянки фібриногену: а-ланцюг Arg-Gly-Asp(RGD) та уланцюг His-His-Leu-Gly-Gly-Ala-Lys-Gln-Ala-Gly-Asp-Val (HHLGAKQAGD V, g400-411). Після того, як було виявлено, що ці фрагменти сповільнюють зв'язування фібриногену з GP ІІв/ІІІа стр уктури, подібні до цих фрагментів, також можуть бути антагоністами. Дійсно, раніше цього винаходу було відкрито, що RGD-подібні антагоністи сповільнюють зв'язування фібриногену з GP ІІв/ІІІа та агрегацію тромбоцитів, наприклад, Ro438857(L.Alig, J.Med. Chem. 1992, 35, 4393) має ІС50 для 0.094mM проти викликаної in vitro тромбіном агрегації тромбоцитів. Деякі з цих агентів також виявили in vivo дію як антитромботичні агенти, в деяких випадках були використані разом з фібринолітичною терапією, наприклад, t-PA або стрептокіназа (J.A.Zablocki, Current Pharmaceutical Design, 1995, 1, 533). Як показано результатами фармакологічних досліджень, описаних нижче, сполуки цього винаходу показують здібність до блокування зв'язування фібриногену з ізольованим GP ІІв/ІІІа (ІС50=0.0002-1.39mМ), сповільнюють агрегацію тромбоцитів in vitro в присутності різних стимуляторів тромбоцитів (0.019-65.0mМ проти тромбіну) і, в подальшому, сповільнюють ex vivo агрегацію тромбоцитів в тваринних моделях. Додатково, ці агенти виявили ефективність на тваринних моделях тромбозів, таку, як показали їх попередники ("Nipecotic Acid Derivatives As Antithrombotic Compounds", номер заявки №08/213772, подана 16 березня 1994p.). Сполуки цього винаходу показують ефективність як антітромботичні агенти завдяки своїй здатності попереджувати агрегацію тромбоцитів. Додатково сполуки цього винаходу сповільнюють інтегрін-зв'язані клітинно-клітинні спайки, вони також можуть використовуватись проти запалення, метастазування ракових клітин та ін. (D.Cox, Drug News&Perspectives 1995, 8,197). Даний винахід стосується сполук, що представлені наступною загальною формулою (І): де А, X, M, R5, R10 та n визначені нижче. Ці сповільнювачі агрегації тромбоцитів можуть бути корисними для лікування зв’язаних з тромбоцитами хвороб, таких як артеріальні та венозні тромбози, гострий інфаркт міокарда, нестійка ангіна, закупорка судин, разом з тромболітичною терапією та ангіопластією, запалення та ряд хвороб, зв’язаних з закупоркою судин. Ці сполуки також корисні як антитромботичні агенти, використані разом з фібринолітичною терапією (наприклад, т-РА або стрептокіназа). Фармаіцевтичні композіції, що включають ці сполуки, також є частиною даного винаходу. Більш детально, даний винахід стосується сполук наступної формули (І): де М - це (СН2)m або піперидин-1-іл; де А вибирається з піперидин-2-іл, піперидин-3-іл, піперидин-4-іл, піперазин-1-іл, піролідин-2-іл, піролідин3-іл, NHR2 або де R9 вибирається з Н, алкіл, CH(NH), СМе(МН) або ацил, переажно R9- водень; де R10 - це H або C(O)N(R 1)YZ де R1 вибирається з Н або циклоалкіл; де R2 вибирається з Н, алкіл або ацил. Переважно R2 - це водень; де R5 -це Н або C(O)NHQ(CHW)rCO2R8; де Q вибирається з СН2, СН-арил, СН-гетероарил, СН-заміщений гетероарил, СН-алкіл; переважно Q - це СН2 , СН - заміщений гетероарил або СН-гетероарил; W вибирається з Н або N(R6)T-R7, переважно W - це Н, коли Q - це СН та N(R6)-T-R7 ,коли Q - це СН2; де R6 вибирається з Н, алкіл або ацил; переважно R6 - це водень, Т вибирається з С(О), C(N-CN) або SO2, переважно Т-це С(О), та R7 вибирається з алкіл, арил, арилалкіл, алкокси або аміноалкіл; та R8 вибирається з Н, алкіл або арилалкіл; переважно R8 – це Н. Де m - це ціле число 1, 2 або 3. Переважно m=1 або 2; де X вибирається з С(О), С(О)О, C(O)NH, CH 2 або SO2; де n - це ціле 1, 2 або 3; де r=0 або 1; де Y вибирається з (СН2)p, CH(R3)(CH2)q , (CH2)q CH(R3), (CH(COR4)CH2)q , (СH2)СНОН або піперидин-3карбонової кислоти; тоді як Y - це (СН2)p та р=2, X не є С(О) або коли Х - це С(О), тоді або R 1 не є Н, або R2 не є Н, та тоді як Y - це (CH(CO2R4)CH2)q X не є С(О) або СН 2; де р=2 або 3; де q=1, 2 або 3. Переважно q=1; де R3 - це алкіл, C2-C8алкеніл, C2-C8алкініл, арил, арилалкіл, або гетероарил; де R4 - це Н, або алкіл, або ціклоалкіл. Переважно R4 - це водень. Де Z - це СО2Н , СО2алкіл, SO3H, РО 3Н2, або 5-тетразол; мається на увазі, що або R5 або R10 - це водень; енантіомер або фармацевтично прийнятна сіль. Переважно, група C(O)NR1YZ зв'язана з атомом вуглецю центрального азациклу в третьому або четвертому положенні (четвертому положенні, коли цикл більший ніж п'ятичленний), але, переважно, в третьому положенні. Як тут мається на увазі, якщо означений алкіл або алкоксигрупа згадуються окремо або як частина заміщеної групи, то вони включають прямі та розгалужені ланцюги, що мають 1-8 атомів вуглецю. Наприклад, алкільні радикали включають метил, етил, пропіл, ізопропіл, н-бутил, ізобутил, вторбутил, т-бутил, н-пентил, 3(2-метил)бутил, 2-пентил, 2-метилбутил, неопентил, н-гексил, 2-гексил, 2-метилпентил. Алкоксильні радикали - це етери, утворені з попередньо описаних прямих або розгалужених ланцюгів алкільних гр уп. Циклоалкільні групи складаються з п'яти-вісьмичленних циклів, переважно з шести- сімичленних циклів. Термін "арил", "гетероарил" або "заміщений гетероарил", як тут мається на увазі, означає ароматичну або гетероароматичну групу, таку як: феніл, нафтил, піридил, тієніл, фураніл або хінолініл, іноді заміщену алкільною групою. Термін "арилалкіл" означає алкільну груп у, заміщену арильною групою. Термін "ацил", як тут мається на увазі, означає органічний радікал, що має 2-6 атомів вуглецю, отриманий від органічної кислоти видаленням гідроксильної групи. Сполуки цього винаходу можуть бути представлені у вигляді фармацевтично прийнятної солі. Фармацевтично прийнятна сіль має таку будову, коли азот 1-піперидина (піролідина, піперазина) протонований неорганічною або органічною кислотою. Представлені неорганічні або органічні кислоти включають соляну, гідробромідну, гідройодидну, перхлоратну, сірчану, азотну, фосфорну, оцтову, пропіонову, бурштинову, малеїнову, фумарову, малонову, винну, лимонну, бензойну, мигдалеву, метансульфонову, гідроксиетансульфонову, бензолсульфонову, щавлеву, 2-нафталінсульфонову, п-толілсульфонову, циклогексансульфонову, саліцилову, са харінову або трифтороцтову. Сполуки цього винаходу представлені в таблиці 1, де "зам." означає місце з' єднання групи C(O)N(R1)YCO2H з центральним азациклом, і де літера "R" після цифри 3 означає абсолютну конфігурацію (за правилами Кана-Інгібіюванняольда-Прелога). Там, де після цифри немає літери - означає рацемічну суміш. Сполуки винаходу, де R5 - це Н, R10 - це C(О)NCR1)YZ, М - це (СН2)m та А - це піперидин-2-іл, піперидин-3іл, піперазин-1-іл, піролідин-2-іл, піролідин-3-іл або NHR2 можна синтезувати, як показано на схемі АА. На цій схемі на алліловий естер ніпекотинатної кислоти (рацемічну суміш або окремий енантіомер) діють 4ппіперидинпропіоновою кислотою на носії в присутності DIC/HOBT та третинного аміну. Потім алліловий естер видаляють за допомогою паладієвого каталізатора, та послідовний процес конденсації відбувається з утворенням кінцевого продукту після омилення триметилсиланоатом калію (наприклад, сполука 1). За аналогією, перегрупування, що базуються на сечовинах та уретанах для третинних амінів (наприклад, сполуки 2 та 3) були проведені реакцією аміну на твердому носії (спирту) з п-нітрофенілхлорформіатом та, потім, з етилніпекотатом (S.M. Hutchins, Tetahedron Lett. 1994,35. 4055) Інтермедіати естерів тризаміщеної 3-амінопропіонової кислот було синтезовано з використанням модифікованої реакції Кневенагеля (Схема AG; E.Profft, J.Pract. Chem. 1965, 30, 18) з подальшою естеріфікацією за Фішером карбонової кислоти (якщо вони недоступні). Ці інтермедіати були отамані з енантіомерним надлишком після розділення рацемічних фенілацетамідів, таких, як AG3 за допомогою пеніцилін-амідази (V.A.Soloshonok, Tetrahedron: Asymetry1995, 6, 1601). Так, небажаний R-енантіомер гідролізується амідазой, тоді як бажаний S-ізомер зберігає фенілацетильну груп у. Розділення також може бути зроблено за допомогою солей (-)-ефедрину або рацемічних тризаміщених 3-N-Boe-амінопропіонових кислот, як викладено (J.A.Zablocki, J.Med. Chem. 1995, 38, 2378). Етилніпекотат та етилізоніпекотат можна знайти в продажу. Синтези 5- та 7- членних аналогів ніпекотамідів (4 та 17, переважно)були виконані твердофазним методом з використанням метилпіролідин-3-карбоксилатних та метилгексагідроазепін-3-карбоксилатних інтермедіатів для аналогів олідин-3-карбоксилат та метилгексагідроазепін-3-ної конверсії АА2 у ААЗ (С хема АА). Метилпіркарбоксилат було отримано, як в публікації (H.Rapoport, J.Org. Chem. 1974, 3, 893). Наприклад, Nбензилгексагідроазепін-2-он було введено в реакцію з літій діізопропіламідом/діетилкарбонатом, продукт потім відновили літійалюмогідридом з утворенням N-бензил-3-гідроксиметилгексагідроазепіну. Бензильну гр упу було еліміновано гидрогенолізом (Н2, Pd-C, метанол), або захищено (ді-т-бутилдикарбонат/гідроксид натрію), спирт окислено триоксидом хрому з утворенням N-Boc-гексагідроазепін-3-карбонову кислоту. Вос-груп у було знято естеріфікацією з використанням НСІ/метанол з утворенням метилгексагідроазепін-3-карбоксилату. Піперазинові аналоги було синтезовано так, як викладено на схемі АВ (S.G. J.Am. Chem Soc. 1988, 110, 6172). Тетразоли (13) були синтезовані з відповідних нітрилів та азидотриметилсилан/дибутиліноксида (Схема AC; S.J.Wittenberger, J.Org. Chem. 1993, 58, 4139). Попередник нітрилу АС2 було синтезовано стандартним амідуванням з 3-амінопропіонітрилу, і відновлено на кінцевій стадії синтезу гідрогенуванням на діоксиді платини (W.J.Hoekstra, J.Med. Chem. 1995, 38, 1582). N-Метилпіперидинові аналоги можуть бути синтезовані за методикою твердофазного пептидного синтезу, що базується на Fmoc-захисті, як це показано на схемі AD (P. Sieber, Tetrahedron Lett. 1987, 28, 6147). Fmocзахисні групи було знято 20% піперидином в диметилформаміді, конденсацію було активовано DIC/HOBT/DMF, кінцеві продукти було видалено з носія за допомогою 95% трифтороцтової кислоти. Сульфонамід 12 було синтезовано, як показано на схемі АЕ. Інтермедіат АЕ1 було ізольовано в дві стадії з 4-піридинетансульфонової кислоти гідрогенуванням/захистом як описано (J.I.deGaw, J.Heterocyclic Chem. 1966, 3, 90), з подальшим хлоруванням тіонілхлоридом за стандартною процедурою (P.J.Hearst, Org. Syn. 1950, 30, 58) з утворенням АЕ2. Інтермедіат АЕ2 було потім переведено в кінцевий продукт за допомогою стандартного синтезу в розчині (WJ.Hoekstra, J. Med. Chem. 1995, 38, 1582). Піперидинілніпекотамід 20 було синтезовано як показано на схемі AF. Естер AF1 було Вос-захищено за допомогою стандартних Boc-ON умов (D.C.Tarbell, Proc. Natl. Acad. Sci. USA 1972, 69, 730), та потім відновлено до відповідного первинного спирту за допомогою DiBAL-H/THF (E.Winterfeldt, Synthesis 1975, 617) з утворенням інтермедіату AF2. Цю сполуку було переведено в відповідний тозилат AF3 за допомогою птозилхлориду (L.F.Awad, Bull. Chem. Soc. Jpn. 1986, 59, 1587). Етилніпекотат потім було алкіловано інтермедіатом AF3 з використанням стандартних умов (бензол/нагрівання; І. Seki, Chem. Pharm. Bull. Jpn. 1970, 18, 1104). Енантіомерно збагачений R-(-)-етиловий естер ніпекотинатної кислоти було виділено хіральним розділенням рацемічної суміши як відповідні солі з D-тартратною кислотою (A.M.Akkerman, Rec. Trav. Chim. Pays-Bas1951, 70, 899). Сполуки цього винаходу представлені в таблиці 1 (та в таблиці 2), де літера "R" після цифри 3 означає абсолютну конфігурацію (за правилами Кана-Інгібіюванняольда-Прелога). Антагоністи винаходу на основі діамінопропіонової кислоти , де R 5 – це C(O)NHQ(CHW)rCO2R8, R10 - це Н, М - це піперидин-1-іл, А – можуть бути синтезовані як показано на схемі АН. Метил-N-a-Zдіамінопропіонат було ацильовано HBTU-активованим АНІ, Z-гр упу було знято гідрогенолізом з утворенням АН2 (для 23 Z-групу залишили), потім отриманий первинний амін було введено в реакцію з потрібним ізоціанатом (або алкілхлорформіатом для 24, алкилсульфонілхлоридом для 25) з утворенням АН3. Вос-групу інтермедіату АН3 було знято НСl, і утворений вторинний амін проацилювали HBTU-активованим АН4 з утворенням АН5. Цей продукт було омилено гідроксидом літію, та Вос-груп у знято НСl з утворенням 22. Антагоністи винаходу на основі біпіперидин-сечовини можуть бути синтезовані як показано на схемі AJ. Інтермедіат AJ1 було синтезовано як показано на схемі AG. AJ1 було ацильовано пнітрофенілхлорформіатом, потім введено в реакцію з Вос-піперидином (для синтезу дивись W. Bondinell, patent application WO 94/14776). Естер AJ2 було омилено гідроксидом літію, та Вос-групу знято НСl з утворенням 27. Інтермедіати заміщених альдегідів піперидинового ряду, такі як АК2, було синтезовано відновленням літійалюмогідридом відповідних метилових естерів нікотинової кислоти (АК1) з подальшим окисненням диоксидом марганцю (Схема АК). Потім альдегіди було переведено в b-амінокислоти як показано на схемі AG. Формамідін AL3 було синтезовано як показано на схемі AL. Амін AL1 було ацильовано етилформіатом як описано М.К.Scott ( J.Med. Chem. 1983, 26, 534). Естер AL2 було омилено 4N НСl (RT, 20 годин) з утворенням 33. Антагоністи на основі тризаміщених (3-амінокислот можуть бути синтезовані як показано на схемі AM. 6-Метилпіридил-b-аміноустер AMI, та продукт конденсації було оброблено НСl з утворенням аміну АМ2. Амін було ацильовано HBTU-активованим АМ4, естер омилено, Вос-групу знято НСl з утворенням 31. Щоб приготувати фармацевтичну композицію цього винаходу, одна або більше сполук формули (І), або їх солі як активного Інгібіюванняредієнту, вони змішуються з фармацевтичним носієм згідно з технологіями формування композицій; ці носії можуть широко варіюватися, в залежності від призначення, наприклад, орального або парентерального, такого як внутрим'язове. Для приготування композиції для орального вживання, будь-яке з звичайних фармацевтичних засобів може бути задіяне. Таким чином, для рідкої оральної форми, такої як: суспензії, еліксири та розчини, придатні носії та доповнювачі включають воду, спирти, масла, гліколі, смакові добавки, консерванти, барвники та ін., для твердої оральної форми, такої як: порошки, капсули, таблетки, придатні носії та доповнювачі включають крохмаль, цукор, розбавлювачі, розрихлювачі, зв'язуючи агенти, либріканти та ін. Завдяки своїй легкості у вживанні, капсули та таблетки представляють найбільш переважну форму для орального дозування, в цьому випадку тверді фармацевтичні носії звичайно використовуються. За бажанням, таблетки покриті сахаром або іншим за стандартними технологіями. Для парентеральних форм носій звичайно включає стерильну воду, хоча інші Інгібіюванняредієнти, наприклад, для збільшення розчинності або консервації, теж можуть бути включені. Також можна приготувати суспензії для ін'єкцій, з відповідним носієм та суспендуючими агентами. Фармацевтичні композиції для цих сполук можуть містити разову дозу, наприклад, таблетки, капсули, порошок, ін'єкції, та інше, певну кількість Інгібіюванняредієнтів, потрібних для ведення ефективної дози як описано вище. Фармацевтичні композиції для цих сполук можуть містити разову дозу, наприклад, таблетки, капсули, порошок, ін'єкції, супозитарії, та інше, від біля 0.03мг до 100мг/кг (бажано 0.1-30мг/кг) та можуть бути дозовані від 0.1-300мг/кг/день (бажано 150мг/кг/день). Дозування можуть бути змінені в залежності від потреби пацієнта, серйозності стану, що лікується та сполуки, що застосовується. Може застосовуватись як щоденне призначення, так і довгострокове дозування.Б Сполуки даного винаходу заважають зв'язуванню фібріногена з GP llв/Іllа, сповільнюють агрегацію тромбоцитів. Внаслідок цього, такі сполуки можуть бути корисними для лікування хвороб, зв’язаних з тромбоцитами, таких як артеріальні та венозні тромбози, гострий інфаркт міокарда, нестійка ангіна, закупорка судин, разом з тромболітичною терапією та ангіопластикою, запалення та ряд хвороб, зв’язаних з закупоркою судин. Рецептор фібріногену (GP llв/Іllа) активується такими стимуляторами, як АДФ, колаген, та за зв'язування з тромбіном відповідають дві різні білкових ділянки фібріногену: a-ланцюг Arg-Gly-Asp (RGD) та gланцюг 400-411. Як показано результатами фармакологічних досліджень, описаних нижче, сполуки цього винаходу показують здібність до блокування зв'язування фібриногену з ізольованим GP llв/Іllа (ІС50=0.00021.39mМ) сповільнюють агрегацію тромбоцитів in vitro в присутності різних стимуляторів тромбоцитів (0.01965.0mМ тромбіну) і, в подальшому, сповільнюють ex vi vo агрегацію тромбоцитів в тваринних моделях. Кінцева ступінь в агрегації тромбоцитів-це зв'язування фібріногена з активовани м глікопротеїном тромбоциту llв/Іllа (GP llв/Іllа), Інгібіюванняібування цього зв'язування представляє наявний антитромботичний підхід. Твердофазний тест in vitro на зв'язування з очищеним глікопротеїном llв/Іllа 96 зразків Іммулон-2 - мікротитрованих чашек (Dynatech-Immulon) заповнили 50мл/зразок RGD-очищеним GP llв/Іllа (ефективна концентрація 0.5-10мг/мл) в 10мл HEPES, 150мл NaCl, 1мМ MgCl2 при рН7.4. Чашку накрили та витримали ніч при 4°С. Розчин GP llв/Іllа видалили, додали 150мл 5% БСА та витримали при кімнатній температурі 1-3 години. Чашку інтенсивно промивають. Отриманий з живої тканини фібріноген (25мл/зразок) при 2х кінцевій концентрації. Чашку накрили та витримали при кімнатній температурі 2-4 години. За 20хв. до кінця інкубації каплю реагенту A (Vecta Stain ABC Horse Radish Peroxidase kit, Vector Laboratories, Inc.) та одну каплю реагенту В додають при перемішуванні до 5мл модифікованого буфера Тірода, залишають. Розчин ліганду видаляють, чашки промивають (5х200мл/зразок) модифікованим буфером Тірода. Реагент Vecta Stain HPRBiotin Avidin (50мл/зразок) додають та витримують при кімнатній температурі 15 хвилин. Розчин Vecta Stain видаляють, чашки промивають (5х200мл/зразок) модифікованим буфером Тірода. Додають розвиваючий буфер (10мл 50мМ цитратно-фосфатного буферу при рН 5.3, 6мг о-фенилендіаміну, 6мл 30% перекису водню, 50мл/зразок) та витримують при кімнатній температурі 3-5 хвилин, потім додають 2N H 2SO4 (50мл/зразок). Знімають поглинання при 490нМ. Результати показано в таблиці lll та IV. In vitro тест на інгібіюванняібування викликаної тромбіном агрегації тромбоцитів Процент агрегації тромбоцитів вираховується як збільшення світлопропускання концентрату, на який подіяли відповідною сполукоюв порівнянні з контрольним концентратом тромбоцитів. Кров людини було отримано від вільних від ліків, нормальних донорів в пробірках, що містять 0.13М цитрату натрію. Збагачену тромбоцитами плазму (ЗТП) збирають центрифугуванням всієї крові при 200g на протязі 10 хвилин при 25°С. Збагачену тромбоцитами плазму (5мл) фільтрують через Sepharose 2B (об'єм 50мл), кількість тромбоцитів приведено до 2*107 тромбоцитів на зразок. До сіликонової кювети додають наступні компоненти: концентрований фільтрат тромбоцитів, буфер Тірода (0.14М NaCl, 0.0027МКСl, 0.012MNaHCO3, 0.76M Na2HPO4, 0.005 5М глюкози, 2мг/мл BSA, 5.012М HEPES при рН7.4) в кількості, еквівалентній 350мл, 50мл 20мМ кальцію та 50мл тестованої сполуки. Агрегацію спостерігають на агрегометрі BIODATA через 3 хвилини після додавання агоністу (тромбін 50мл на 1 зразок). Результати показано в таблиці lll та IV. Таблиця lll Результати in vitro зв 'язування агрегація Сполука фібріногену тромбоцитів * № % Інгібіюв ання % Інгібіюв ання ІС50(mМ) ІС50(mМ) (50цМ) (50цМ) 1 95.0 0.003 83.0 3.6 2 93.0 0.027 95.7 54.0 3 81.0 нт 26.2 >100 4 89.9 0.121 81.0 26.0 5 89.0 0.012 100 10.0 6 90.7 0.197 71.2 73.0 7 100 0.006 75.6 2.4 8 93.0 0.332 94.8 65.0 9 99.0 0.002 90.9 0.37 10 91.3 0.019 85.0 1.6 1 79.6 0.004 99.2 1.55 12 97.0 0.025 88.0 15.5 13 95.0 1.39 67.0 25.5 14 99.0 0.004 91.0 0.91 15 100 0.0091 92.2 1.9 16 100 0.0005 94.0 0.028 17 96.0 0.005 89.6 0.45 18 100 0.0002 100 0.019 19 99.0 0.021 92.1 0.079 20 99.0 0.0007 89.7 37.0 21 100 0.0005 100 0.060 * Тромбін-індуков ана агрегація гель-фільтров аних тромбоцитів Таблиця IV Результати дослідів in vitro зв 'язування тромбоцитна Сполука фібріногену агрегація * № % Інгібіюв ання ІС50 % Інгібіюв ання ІС50 (50мкМ) (мкМ) (50мкМ) (мкМ) 22 100 0,0007 94,0 0,046 23 100 0,0003 97,0 0,027 24 100 0,0004 100 0,018 25 100 0,0003 97,0 0,007 26 100 0,0003 97,0 0,016 27 100 0,0006 100 0,45 28 100 0,0002 100 0,17 29 100 0,068 100 42 30 100 0,0008 100 0,19 31 100 0,0003 100 0,045 32 100 0,0004 100 0,020 33 100 0,0007 100 0,30 * Агрегація гель-фільтров аних опосередкована тромбіном. тромбоцитів, Ex vi vo вивчення на собаках Дорослих дворняг (8-13кг.) ввели у стан наркотичного сну за допомогою пентобарбіталу натрію (35мг/кг., інтравенозно) та підтримували їх життєдіяльність за допомогою штучного дихання. Артеріальний кров'яний тиск та швидкість серцебиття вимірювали за допомогою катетерного наконечнику Міллара як датчик тиску, який вставлено у стегнову артерію. Інший датчик Міллера розташували у лівому шлуночку (ЛШ) через сонну артерію для вимірювання кінцевого діастолічного тиску у лівому шлуночку та індексів міокардіальних скорочень. Електрокардіограму реєстрували за допомогою електродів, які приєднали безпосередньо до тіла. Катетори розташували у стегновій артерії та вені, щоб відбирати проби крові та вводити ліки, відповідним чином. Відклики безперервно контролювали із використанням системи обробки даних Modular Instruments. Проби артеріальної крові (5-9мл) додали до пробірок., що містили 3,8% розчин цитрату натрію для приготування плазми, що збагачена тромбоцитами (ПЗТ) та визначення впливу на параметри коагуляції: час життя протромбіну (ЧП) та час життя частково активованого тромбопластину (ЧЧАТ). Ізольовані проби крові (1,5мл) додали у розчин ЕДТА для визначення кількості гематокритів та клітин (тромбоцитів, червоних та білих кров'яних клітин). Еталон часу кровотоку отримали з поверні щоки використовуючи розрізаючий пристрій та фільтрувальний папір Ватмана (Whatman). Агрегація плазми, що збагачена тромбоцитами була здійснена із використанням BioData агрегометра. Для агрегації усієї крові використали імпедансний агрегометр Chronolog. Час життя протромбіну та час життя частково активованого тромбопластину вимірювали з допомогою приладу BioData чи аналізатора коагуляції ACL 3000+. Кількість клітини підраховували на приладі Sysmex K-1000. Сполуки розчинили у малий кількості диметилформаміду (ДМФА) та розбавили насиченим водним розчином солі, щоб кінцева концентрація диметилформаміду складала 10%. Сполуки вводили інтравенозно з допомогою інфузіонного насосу Harward. Дози вводили через кожні 15 хвилин із постійною швидкістю 0,33мл/хв. Дані реєстрували після введення кожної дози та через 30 хвилинні інтервали після закінчення введення ліків. Ліки вводили орально у вигляді водного розчину із допомогою шприца. Досліджувані сполуки викликали вищезазначене інгібіювання ex vi vo тромбоцитної агрегації. Таким чином, в крові ці сполуки інгібіювали колаген-стимульовану (чи АДФ-стимульовану) агрегацію у дозах 0,1-10мг/кг, при цьому треба зазначити, що інгібіювання колагену стимульовало тромбоцитний АТФ відклик. В плазмі, що збагачена тромбоцитами, ці сполуки також інгібіювали колаген-стимульовану тромбоцитну агрегацію, при цьому активність проявлялася в інтервалі 0,1-10мг/кг. Сполуки не мають виміряного гемодинамічного ефекту у дозах аж до 1мг/кг інтравенозно. Ліки викликають збільшення еталонного часу кровотоку при концентрації 0,11мг/кг із наступним швидким поверненням до початкового рівня. Ніякого ефекту коагуляції (час життя протромбіну чи час життя частково активованого тромбопластину) не спостерігалося на протязі обробки та тромбоцити, білі та червоні кров'яні тіла залишалися без змін при довільних дозах цих сполук. Із отриманих результатів можна зробити висновок, що сполуки є високо ефективними інгібіторами тромбоцитної агрегації ex vi vo (антагоністи колаген- та АДФ-стимульованого шляху агрегації) при інтравенозному введенні їх в межах 0,1-1мг/кг чи 1-10мг/кг орально (таблиці V та VI). Ан тиагрегативний ефект супроводжувався збільшенням часу кровотоку при більших дозах. Ані гемодинамічного, ані гематологічного ефектів при цьому не спостерігалося. Таблиця V Ex vivo в ивчення на собаках Сполука Інтрав енозне введення Оральне вв едення Доза, Трив алість*, Доза, Трив алість*, № мг/кг хв . мг/кг хв . 15 1 30 10 120 16 0,1 60 1 60 17 0,3 3 >180 18 од зо 1 150 19 1 зо 10 90 21 0,3 150 1 180 * Відпов ідає тривалості >50% інгібіюв ання колагенчи АДФ-опосередков аної ех vivo тромбоцитної агрегації. Таблиця VI Ex vivo в ивчення на собаках Сполука Інтрав енозне введення Оральне вв едення Доза, Трив алість*, Доза, Трив алість*, № мг/кг хв . мг/кг хв . 22 0,3 180 3 60 23 од 60 1 180 24 0,3 3 150 25 0,3 90 3 120 26 0,3 30 3 60 27 0,3 3 60 28 0,3 60 3 120 29 0,3 3 120 30 0,3 105 3 180 31 0,3 120 3 >180 32 0,3 60 3 180 * Відпов ідає тривалості >50% інгібіюв ання колагенопосередкованої ex viv o тромбоцитної агрегації. Сполуки 16 та 18 показують ефективність у артеріовенозній моделі шунта тромбоза для собаки, при чому вона залежить від дози (метод описан у "Nipecotic Acid Derivatives As Antithrombotic Cjmpounds", №08/213772, март 16, 1994г.). Наприклад, сполука 16 інгібує утворення тромбозу при 10, 30 та 100мкг/кг/хв кумулятивних дозах при введенні інтравенозно (75%, 37%, 12% від загальної маси тромбів відповідно). Сполука 18 інгібує утворення тромбів при 3, 10 та 30мкг/кг/хв. кум улятивних дозах при введенні інтравенозно (82%, 41%, 12% від загальної маси тромбів відповідно). Приклади Захи щені амінокислоти придбали у компанії Aldrich Chemical чи Bachem Bioscience Inc. 2-Хлортритильний смоляний сорбент та сорбент Ванга (Wang) отримали від компанії Novabiochem Corp. Енатіомерно збагачені етилові естери циклоалкіліден-3-карбонової кислоти були виділені розділенням рацемічних сумішей за відомою методикою (А.М.Akkerman, Rec. Trav. Chim. Pays-Bas 1951, 70, 899). Усі інші хімічні реактиви були придбані в компанії Aldrich Chemical Company, Inc. Кінцеві продукти, що одержали у вигляді кислотноадитивних солей можуть бути переведені у вільні основи за допомогою основної іон-обмінної хроматографії. 1 Н-ЯМР спектри високої резолюції реєстрували на спектрометрі Bracer АС-360 при 360МГц та константи спін спінової взаємодії вимірювалися у Гц. Температури топлення визначалися на приладі Mel-Temp II та не надалі не уточнювалися. Мікроаналізи були проведені в компанії Robertson Microlit Laboratries, Inc., Madison, New Jersey. У ти х випадках, коли продукт отримали у вигляді солі, вільну основу одержували загальновідомими методами, наприклад, до солі додавали основу із подальшою очисткою отриманої цільової органічної основи. У прикладах, що наведені тут., надалі буде використовуватися така абревіатура. Вn чи Bzl= бензил; Вое= t-бутоксикарбоніл; ВОС-ON=2-(1-бутоксикарбонілоксіміно)-2-фенілацетоштрил; ВОР-Сl= біс(2-оксо-3-оксазолідиніл)фосфініл хлорид; СР= сполука; DCE= 1,2-дихлоретан; DCM= дихлорметан; DIBAL-H= діізобутилалюміній гідрид; DIC= діізопропілкарбодіімід; DIEA= діізопропілетиламін; DMAP= 4-диметиламінопіридин; DMF= N,N-диметилформамід; EDC= етил диметиамінопропілкарбодіімід; EDTA= е тилендіамінтетраоцтова кислота; Et2O= діетиловий етер; HBTU= 2-(1H-бензотріазол-1-іл)-1,1,3,3-тетраметилсечовини гексафторфосфат; НОВТ= гідроксибензотріазол; і-Рr= ізопропіл; KOTMS= калію триметилсиланоат; NMM= N-метилморфолін; Nip= ніпекотил (рацемічний по 3-положенню); NT= не тестований; РРТ= осадження; PTSA= р-толуенсульфонова кислота; RT= кімнатна температура; TFA= трифтороцтова кислота; ТМSN3= азидотриметилсилан; Z= бензилоксикарбоніл. Аліл 3-(4-піперидин)пропіонат·НСl (АА1 прекурсор) До суміші 3-(4-піридил)акрилової кислоти (10,0г, 0,066моль) та водного розчину соляної кислоти (2,0N, 50мл) в атмосфері азоту додали оксид платини (IV) (0,54г). Цю суміш гідрогенезували при 50атм. та кімнатній температурі на протязі 21год., потім профільтрували через целіт та суміш випаровували, що дало гідрохлорид 3-(4-піперидил)пропіонової кислоти у вигляді білого порошку (12,9г, 99%). Цей порошок обробили аліловим спиртом (50мл) та гріли при 50°С на протязі 2год. Потім розчин охолодили до кімнатної температури, випарували до об'єму приблизно 10мл та розбавили діетиловим етером (250мл). Цільовий осад відфільтрували та промили діетиловим етером, що дало білий порошок (14,5г, 94%): 1Н-ЯМР (ДМСО-D6) d 8,79,1 (м, 2Н), 5,9 (м., 1H), 5,25 (д.д., J= 7,15, 2Н), 4,53 (д, J= 4, 2Н), 3,21 (д., J= 8, 2Н), 2,74 (т., J= 7, 2Н), 2,35 (т., J= 4, 2H), 1,72 (д., J= 8, 2Н), 1,5 (м., 3Н), 1,3 (м., 2Н); мас-спектр: m/e 198 (МH+). Метил (S)-3-аміно-3-(З-піридил)пропіонат·2НСІ (AG5) Фенілацетамід інтермедіату AG3 синтезували за відомою методикою, що показана на схемі AG (Е.Profft, J.Prakt. Chem. 1965, 30, 18). Суміш AG1 (0,47моль), етилового спирту (100мл), ацетату амонію (0,47моль) та малонової кислоти (0,70моль) нагрівали із оберненим холодильником на протязі 6год., потім охолодили суміш та відфільтрували. Білу тверду речовину промили етиловим., а потім метиловим спиртом та висушили. Отриману тверду речовину розчинили в суміші 2:1= ацетон:вода (360мл), обробили триетиламіном (0,72моль) та фенілацетилхлоридом (0,36моль), та перемішували реакційну суміш на протязі 22год. Суміш випарували та залишок розчинили у воді (500мл), рН розчину довели до 12 (IN розчин NaOH). Потім водну фазу підкислили до рН2 (конц. водний розчин соляної кислоти), екстрагували діетиловим етером та випарували, що дало речовину у вигляді білої піни. Цю піну очистили хроматографією на силікагелі (10% МеОН/дихлорметан), що дало AG3. Розчин AG3 (0,22моль) у воді (600мл) при кімнатній температурі довели до рН7,5 за допомогою КОН (3,0N) та обробили пеніцилін амідазою (91520 одиниць, Sigma). Цю суміш перемішували на протязі 47год., потім підкислили до рН1 за допомогою водного розчину соляної кислоти (конц.) профільтрували крізь целіт. Фільтрат екстрагували діетиловим етером (тричі по 300мл), сконцентрували у вакуумі та обробили МеОН/ конц. NH4OH (9:1). Отриманий розчин хроматографували на силікагелі (елюент дихлорметан/МеОН/ NH4OH=78/18/4), що дало (S)-3-фенілацетамідо-3-(3-піридил)пропіонової кислоти амонієву сіль (19,5г, 58%). Цей продукт обробили водним розчином соляної кислоти (6,0N, 292мл), гріли із оберненим холодильником на протязі 5год., охолодили до кімнатної температури та екстрагували діетиловим етером (тричі по 200мл). рН водної фази довели до 12, сконцентрували у вакуумі та отриману тверду речовину обробили обробили МеОН (двічі по 300мл). Цей розчин випаровували, що дало приблизно 14г натрієвої солі. Цю сіль обробили МеОН (500мл), 2,2-диметоксипропаном (44мл) та НСІ (4N розчин у діоксані, 84мл) та перемішували на протязі 90год. при кімнатній температурі. Цю суміш відфільтрували та фільтрат сконцентрували у вакуумі. Отриману не зовсім білу тверду речовину обробили діетиловим етером (двічі по 150мл) та висушили, що дало цільову сполуку AG5 (16,7г., 96% ее) у вигляді білої аморфної твердої речовини. Приклад 1 N-3-(4-піперидинпропіоніл)ніпекотил(3-аміно-3-феніл )пропіонової кислоти трифторацетат (1) У сосуд із термостійкого скла ємністю 25мл в атмосфері азоту помістили 2-хлортритил хлоридний смоляний сорбент (0,24г, 0,36 мілімоль, Novabiochem) та диметилформамід (5мл). Смоляний сорбент струшували в атмосфері азоту до набухання та зникнення надлишку диметилформаміду. Смоляний сорбент обробили диметилформамідом (5мл), діізопропілетиламіном (0,31мл, 5 еквіваленти) та аліл 3-(4піперидил)пропіонатом·НСІ (0,20г, 2,4 еквіваленти) у такій послідовності та струшували на протязі 8 год. Отриманий темно-зелений розчин видалили та смоляний сорбент промили диметилформамідом (тричі по 5мл), водним розчином диметилформаміду (25%, тричі по 5мл), тетрагідрофураном (тричі по 5мл), дихлорметаном (тричі по 5мл) та діетиловим етером (5мл). Смоляний сорбент наситили 1,2-дихлоретаном (5мл) до набухання та обробили сумішшю тетрабутиламонію фториду гідрата (0,28г, 3 еквіваленти), азидотриметилсиланом (0,38мл, 10 еквіваленти), тетракіс(трифенілфосфін)паладію (0,084г, 20 мольних %) та 1,2-дихлоретаном (5мл). Смоляний сорбент струшували на протязі 15год. та розчин морквяного кольору відділили. Смоляний сорбент промили дихлорметаном (тричі по 5мл), диметилформамідом (тричі по 5мл), тетрагідрофураном (тричі по 5мл) та діетиловим етером (5мл). Смоляний сорбент наситили диметилформамідом (5мл) до набухання та обробили діізопропілетиламіном (0,18мл, 3 еквіваленти), алілніпекотат гідрохлоридом (0,17г, 3 еквіваленти), діізопропілкарбодіімідом (0,17мл, 3 еквіваленти) та гідроксибензотріазолом (1мг). Смоляний сорбент струшували на протязі 15год. та розчин відділили. Смоляний сорбент промили диметилформамідом (тричі по 5мл), водним розчином диметилформаміду (25%, тричі по 5мл), тетрагідрофураном (тричі по 5мл), дихлорметаном (тричі по 5мл) та діетиловим етером (5мл). Смоляний сорбент наситили 1,2-дихлоретаном (5мл) до набухання та обробили сумішшю тетрабутиламонію фториду гідрата (0,28г, 3 еквіваленти), азидотриметилсиланом (0,38мл, 10 еквіваленти), тетракіс(трифенілфосфін)паладію (0,084г, 20 мольних %) та 1,2-дихлоретаном (5мл). Смоляний сорбент струшували на протязі 15год. та розчин морквяного кольору відділили. Смоляний сорбент промили дихлорметаном (тричі по 5мл), диметилформамідом (тричі по 5мл), тетрагідрофураном (тричі по 5мл) та діетиловим етером (5мл). Смоляний сорбент наситили диметилформамідом (5мл) та обробили діізопропілетиламіном (0,18мл, 3 еквіваленти), метил D,L 3-аміно-3-фенілпропіонат гідрохлоридом (0,23г, 3 еквіваленти) діізопропілкарбодіімідом (0,17мл, 3 еквіваленти) та гідроксибензотріазолом (1мг). Смоляний сорбент струшували на протязі 17год. та розчин відділили. Смоляний сорбент промили диметилформамідом (тричі по 5мл), водним розчином диметилформаміду (25%, тричі по 5мл), тетрагідрофураном (тричі по 5мл), дихлорметаном (тричі по 5мл) та діетиловим етером (5мл). Смоляний сорбент наситили тетрагідрофураном (5мл) до набухання та обробили розчином калію триметилсиланоату (0,23г, 10 еквіваленти) та тетрагідрофураном (2мл). Смоляний сорбент струшували на протязі 18год. та розчин відділили. Смоляний сорбент промили диметилформамідом (тричі по 5мл), сумішшю оцтова кислота/тетрагідрофуран =1/1 (двічі), водним розчином диметилформаміду (25%, тричі по 5мл), тетрагідрофураном (тричі по 5мл), дихлорметаном (тричі по 5мл) та діетиловим етером (5мл). Смоляний сорбент обробили сумішшю трифтороцтова кислота/ дихлорметан (1/1, 10мл) струшували на протязі 15 хвилин та відділили отриманий червоного кольору розчин. Цей розчин випарували та одержану олію обробили діетиловим етером (тричі по 5мл) та висушили осад. Це дало сполуку 1 у вигляді безбарвної склоподібної речовини (0,11г): 1Н-ЯМР (ДМСО-D6) d 8,6 (м., 1Н), 8,42 (д., J= 7, 1H), 8,2 (м., 1H), 7,3 (м., 3Н), 7,2 (м., 2Н), 5,18 (д., J= 6, 1H), 4,3 (м., 1H), 3,7 (м., 1Н), 3,2 (м., 3Н), 2,8 (м., 2Н), 2,6 (м., 2Н), 2,3 (м., 5Н), 1,1-1,9 (м., 11Н); мас-спектр: m/е 416 (МН+). Використовуя подібний загальний метод твердофазного синтезу, який описано у Прикладі 1, були синтезовані сполуки у вказаних прикладах згідно зі схемою АА, як зазначено в окремих прикладах. Приклад 2 N-3-(4-ппіперидинметиламінокарбоніл)ніпекотил(3-аміно-2-метил)пропанової кислоти трифторацетат (2) Сполука 2 була одержана за схемою АА. Адсорбований на смоляному сорбенті 4-піперидинметиламін (0,36мілімоль) наситили 1,2-дихлоретаном (5мл) до набухання та обробили р-нітрофенілхлорформіатом (0,36мілімоль) та діізопропілетиламіном (0,36мілімоль), потім струшували на протязі 1год. та розчинник видалили. Смоляний сорбент промили (див. Приклад 1) наситили 1,2-дихлоретаном (5мл) до набухання та обробили аліл ніпекотат гідрохлоридом (0,36мілімоль) та діізопропілетиламіном (0,72мілімоль), потім струшували на протязі 16год. Розчинник видалили та смоляний сорбент промили (див. Приклад 1), аліловий естер перетворили на відповідну кислоту (див. Приклад 1). Смоляний сорбент наситили диметилформамідом (5мл) до набухання та кислоту ввели в реакцію із метил 3-аміно-2-метилпропіонатом (0,36мілімоль), та далі діяли за методикою, описаною в Прикладі 1. Сполука 2 була виділена у вигляді безбарвної склоподібної речовини (0,11г): 1Н-ЯМР (CD3OD) d 3,9 (м., 2Н), 3,2 (м., 4Н), 3,10 (д., J= 7, 2Н), 2,9 (м., 3Н), 2,6 (м, 2Н), 2,3 (м., 1H), 1,9 (м., 4Н), 1,7-1,9 (м, 5Н), 1,3-1,5 (м., 5Н), 1,11 (д., J= 7, 3Н); мас-спектр: m/е 355 (МH+). Приклад 3 N-3-(4-піперидинметилоксикарбоніл)ніпекотил-D-аспартамової кислоти a-метилового естера трифторацетат (2) Сполука 3 була одержана за схемою АА. Адсорбований на смоляному сорбенті 4-піперидинметанол (0,36мілімоль) наситили 1,2-дихлоретаном (5мл) до набухання та обробили р-нітрофенілхлорформіатом (0,36мілімоль) та діізопропіл етил аміном (0,36мілімоль), потім струшували на протязі 1год. та розчинник видалили. Смоляний сорбент промили (див. Приклад 1) наситили 1,2-дихлоретаном (5мл) до набухання та обробили аліл ніпекотат гідрохлоридом (0,36мілімоль) та діізопропілетиламшом (0,72мілімоль), потім струшували на протязі 16год. Розчинник видалили та смоляний сорбент промили (див. Приклад 1), аліловий естер перетворили на відповідну кислоту (див. Приклад 1). Смоляний сорбент наситили диметилформамідом (5мл) до набухання та кислоту ввели в реакцію із H-D-Asp(Obn)-OMe (0,36мілімоль), та далі діяли за методикою, описаною в Прикладі 1. Сполука 3 була виділена у вигляді жовтої склоподібної речовини (0,019г): 1 Н-ЯМР (CD3OD) d 4,8 (м., 2Н), 3,9 (м., 3Н), 3,70 (д., J= 9, 4Н), 3,39 (с., 3Н), 3,3 (м., 2Н), 2,9 (м., 4Н), 2,8 (м., 2Н), 1,9 (м., 4Н), 1,7 (м., 2Н), 1,4 (м., 4Н); мас-спектр: m/е 400 (МH+). Приклад 4 N-3-(4-піперидинпропіоніл)піролідин-3-карбоніл-[3-аміно-3-(4-толіл)]пропіонової кислоти трифторацетат (4) Сполука 4 була одержана за схемою АА. Інтермедіат АА2 (0,36мілімоль) наситили 1,2-дихлоретаном (5мл) до набухання та обробили піролідин-3-карбоксилат гідрохлоридом (0,36мілімоль), діізопропілкарбодіімідом (0,72мілімоль) та діізопропілетиламіном (0,72мілімоль), потім струшували на протязі 16год. Розчинник видалили та смоляний сорбент промили (див. Приклад 1), а метиловий естер перетворили у відповідну кислоту взаємодією із калієм триметилсиланоатом (див. Приклад 1). Смоляний сорбент наситили диметилформамідом (5мл) до набухання та кислоту ввели в реакцію із метил 3-аміно-3-(4-толіл)пропіонатом (0,36мілімоль), та далі діяли за методикою, описаною в Прикладі 1. Сполука 4 була виділена у вигляді безбарвної склоподібної речовини (0,081г): 1Н-ЯМР (CD3OD) d 7,19 (д., J= 5, 2Н), 7,10 (д., J= 5, 2H), 5,31(д.д., J= 3, 10; 1Н), 3,6 (м., 4Н), 3,3 (м., 2Н), 2,9 (м, 4Н), 2,7 (м., 2Н), 2,3 (м., 2Н), 2,1 (м., ЗН), 1,9 (м., 4Н), 1,6 (м., 4Н), 1,3 (м., 4Н); мас-спектр: m/е 416 (МH+). Приклад 5 N-3-(4-піперидинпропіоніл)ізоніпекотил(3-аміно-3-метил)пропіонової кислоти трифторацетат (5) Сполука 5 була одержана за схемою АА. Інтермедіат АА2 (0,36мілімоль) наситили 1,2-дихлоретаном (5мл) до набухання та обробили етилізоніпекотатом (0,36мілімоль), діізопропілкарбодіімідом (0,72мілімоль) та діізопропілетиламіном (0,72мілімоль), потім струшували на протязі 16год. Розчинник видалили та смоляний сорбент промили (див. Приклад 1), а етиловий естер перетворили у відповідну кислоту взаємодією із калієм триметилсиланоатом (див. Приклад 1). Смоляний сорбент наситили диметилформамідом (5мл) до набухання та кислоту ввели в реакцію із метил 3-аміно-3-метилпропіонатом (0,36мілімоль), та далі діяли за методикою, описаною в Прикладі 1. Сполука 5 була виділена у вигляді безбарвної склоподібної речовини (0,033г): 1H-ЯМР (CD3OD) d 4,5 (м., 1H), 4,2 (м., 1H), 3,9 (м, 1H), 3,3 (м., 2Н), 3,2 (м., 3Н), 3,1 (м., 1Н), 2,9 (м., 3Н), 2,7 (м., 2Н), 2,4 (м., 2Н), 2,0 (м, 2Н), 1,7 (м., 2Н), 1,5 (м., 6Н), 1,3 (м., 2Н), 1,15 (д., J= 9, 3Н); мас-спектр: m/e 354 (МH+). Приклад 6 (6) N-3-(4-піперидинпропіоніл)ізоніпекотил[3-аміно-3-(4-карбоксифеніл)]пропіонової кислоти трифторацетат Сполука 6 була одержана за схемою АА. Інтермедіат АА2 (0,36мілімоль) наситили 1,2-дихлоретаном (5мл) до набухання та обробили етилізоніпекотатом (0,36мілімоль), діізопропілкарбодіімідом (0,72мілімоль) та діізопропілетиламіном (0,72мілімоль), потім струшували на протязі 16год. Розчинник видалили та смоляний сорбент промили (див. Приклад 1), а етиловий естер перетворили у відповідну кислоту взаємодією із калієм триметилсиланоатом (див. Приклад 1). Смоляний сорбент наситили диметилформамідом (5мл) до набухання та кислоту ввели в реакцію із метил 3-аміно-3-(4-карбосифеніл)пропіонатом (0,36мілімоль), та далі діяли за методикою, описаною в Прикладі 1. Сполука 6 була виділена у вигляді жовтовато-брунатної склоподібної речовини (0,034г): 1H-ЯМР (CD3OD) d 7,9 (м., 3Н), 7,43 (д., J= 5, 2Н), 5,4 (м., 1Н), 4,5 (м., 1H), 4,0 (м., 1H), 3,3 (м., 4Н), 3,1 (м., 1H), 2,9 (м., 2Н), 2,7 (м., 2Н), 2,6 (м., 1Н), 2,5 (м., 4Н), 2,0 (м, 2Н), 1,2-1,9 (м., 10Н); мас-спектр: m/e 460 (МH+). Приклад 7 N-3-(4-N-метилпіперидинпропіоніл)ніпекотил-3-амінопропіонової кислоти трифторацетат (7) Сполука 7 була одержана за схемою AD. Адсорбований на смоляному сорбенті Fmoc-b-Ala (1мілімоль) обробили сумішшю 20% піперидин/диметилформамід (10мл), потім струшували на протязі 2год. та розчинник видалили. Смоляний сорбент промили диметилформамідом., наситили диметилформамідом (10мл) до набухання та обробили Fmoc-ніпекотовою кислотою (1мілімоль), діізопропілкарбодіімідом (2мілімоль) та діізопропілетиламіном (1мілімоль). Смоляний сорбент струшували на протязі 16год., розчинник видалили та смоляний сорбент промили диметилформамідом та дихлорметаном. Смоляний сорбент обробили сумішшю 20% піперидин/диметилформамід (10мл) на протязі 2год., розчинник видалили та смоляний сорбент промили диметилформамідом. Смоляний сорбент наситили диметилформамідом (10мл) до набухання, обробили 4-Nметилпіперидинпропіоновою кислотою (1мілімоль), діізопропілкарбодіімідом (2мілімоль) та діізопропілетиламіном (1мілімоль) та струшували на протязі 16год. Розчинник видалили та смоляний сорбент промили диметилформамідом та дихлорметаном. Смоляний сорбент обробили 95% трифтороцтовою кислотою та потім трифтороцтову кислоту випаровували, що дало сполуку 7 у вигляді безбарвного порошку (0,26г): 1Н-ЯМР (CDCI3) d 4,4 (м., 1H), 3,7 (м., 1H), 3,4 (м., 1H), 3,2 (м., 1H), 3,1 (м., 1Н), 2,7 (м, 2Н), 2,3 (м., 6Н), 2,21 (с., 3Н), 1,9 (м., 4Н), 1,3-1,8 (м., 10Н); мас-спектр: m/е 354 (МH+). Приклад 8 N-3-(4-піперидинпропіоніл)ніпекотил-4-оксоніпекотової кислоти трифторацетат (8) Сполука 8 була одержана за схемою АА. Інтермедіат АА2 (0,36мілімоль) наситили 1,2-дихлоретаном (5мл) до набухання та обробили етилніпекотатом (0,36мілімоль), діізопропілкарбодіімідом (0,72мілімоль) та діізопропілетиламіном (0,72мілімоль), потім струшували на протязі 16год. Розчинник видалили та смоляний сорбент промили (див. Приклад 1), а етиловий естер перетворили у відповідну кислоту взаємодією із калієм триметилсиланоатом (див. Приклад 1). Смоляний сорбент наситили диметилформамідом (5мл) до набухання та кислоту ввели в реакцію із метил 4-оксоніпекотатом (0,36мілімоль), та далі діяли за методикою, описаною в Прикладі 1. Сполука 8 була виділена у вигляді безбарвної склоподібної речовини 0,04г): 1Н-ЯМР (ДМСО-ОD6) d 8,5 (м., 1H), 8,2 (м., 1Н), 6,5 (м., 1H), 4,3 (м., 1H), 3,4-3,8 (м., 4Н), 3,2 (м., 2Н), 3,0 (м., 1H), 2,8 (м., 2Н), 2,2-2,6 (м., 6Н), 1,8 (м., 2Н), 1,1-1,7 (м., 11H); мас-спектр: m/e 394 (МН+). Приклад 9 N-3-(4-піперидинпропіоніл)ніпекотил[3-аміно-3-(2-метилсилілетиніл)]пропіонової кислоти трифторацетат (9) Сполука 9 була одержана за схемою АА. Інтермедіат АА2 (0,36мілімоль) наситили 1,2-дихлоретаном (5мл) до набухання та обробили етилніпекотатом (0,36мілімоль), діізопропілкарбодіімідом (0,72мілімоль) та діізопропілетиламіном (0,72мілімоль), потім струшували на протязі 16год. Розчинник видалили та смоляний сорбент промили (див. Приклад 1), а етиловий естер перетворили у відповідну кислоту взаємодією із калієм триметилсиланоатом (див. Приклад 1). Смоляний сорбент наситили диметилформамідом (5мл) до набухання та кислоту ввели в реакцію із метил 3-аміно-3-(2-триметилсилілетиніл)пропіонатом (0,36мілімоль) (методику синтезу див, J.Zablocki, J.Med. Chem., 1995, 38, 2378) та далі діяли за методикою, описаною в Прикладі 1. Сполука 9 була виділена у вигляді жовтої склоподібної речовини (0,12г): 1Н-ЯМР (CD3OD) d 3,8 (м., 1H), 3,2-3,4 (м., 4Н), 2,9 (м., 3Н), 2,7 (м., 2Н), 2,3-2,5 (м., 2Н), 1,9 (м., 4Н), 1,1-1,9 (м, 13Н), 0,0 (с., 9Н); мас-спектр: m/e 436 (МH+). Приклад 10 N-3-(6-аміокапроіл)ніпекотил-3-аміно-3-(3-піридил)пропіонової кислоти трис трифторацетат (10) Сполука 10 була одержана за схемою АА. Смоляноний сорбент на якому адсорбована амінокапрнова кислота (0,36мілімоль) наситили 1,2-дихлоретаном (5мл) до набухання та обробили етилніпекотатом (0,36мілімоль), діізопропілкарбодіімідом (0,72мілімоль) та діізопропілетиламіном (0,72мілімоль), потім струшували на протязі 16год. Розчинник видалили та смоляний сорбент промили (див. Приклад 1), а етиловий естер перетворили у відповідну кислоту взаємодією із калієм триметилсиланоатом (див. Приклад 1). Смоляний сорбент наситили диметилформамідом (5мл) до набухання та кислоту ввели в реакцію із метил 3аміно-3-(3-піридил)пропіонатом (0,36мілімоль), та далі діяли за методикою, описаною в Прикладі 1. Сполука 10 була виділена у вигляді безбарвної склоподібної речовини (0,008г): 1Н-ЯМР (ДМСО-D6) d 8,6 (м., 2Н), 8,1 (с., 1H), 7,0-7,7 (м., 5Н), 5,15 (т., J= 3, 1H), 4,4 (м., 1Н), 4,1 (м., 1H), 3,7 (м., 2Н), 3,1 (м., 1H), 2,7 (м., 4Н), 2,5 (м., 1H), 2,3 (м., 2Н), 1,2-1,9 (м., 11Н); мас-спектр: m/е 391 (MH+). Дані елементного аналізу для С20Н30NО 4·3ТТА·2Н2О (768,60). Розраховано: С, 40,63; Н, 4,85; N, 7,29; F, 22,25. Знайдено: С, 40,81; Н, 4,70; N, 6,12; F, 23,83. Приклад 11 N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил(3-аміно-2-гідрокси)пропіонової кислоти трифторацетат (11) Сполука 11 була одержана за схемою АА. Інтермедіат АА2 (0,36мілімоль) наситили 1,2-дихлоретаном (5мл) до набухання та обробили етил R-ніпекотатом (0,36мілімоль), діізопропілкарбодіімідом (0,72мілімоль) та діізопропілетиламіном (0,72мілімоль), потім струшували на протязі 16год. Розчинник видалили та смоляний сорбент промили (див. Приклад 1), а етиловий естер перетворили у відповідну кислоту взаємодією із калієм триметилсиланоатом (див. Приклад 1). Смоляний сорбент наситили диметилформамiдом (5мл) до набухання та кислоту ввели в реакцію із метил 3-аміно-2-гідроксипропіонатом (0,36мілімоль), та далі діяли за методикою, описаною в Прикладі 1. Сполука 11 була виділена у вигляді рожевої склоподібної речовини (0,05г): 1Н-ЯМР (ДМСО-D6) d 8,5 (м., 1H), 8,2 (м., 1H), 7,6 (м., 1H), 4,0-4,4 (м., 2Н), 3,7 (м., 1Н), 3,2 (м., 3Н), 2,8 (м., 3Н), 2,6 (м., 1H), 2,1-2,3 (м., 3Н), 1,8 (м, 4Н), 1,0-1,4 (м., 10Н); мас-спектр: т/е 356 (МH+). Приклад 12 N-3-(4-піперидинетансульфоніл)ніпекотил-3-амінопропіонової кислоти гідро хлорид (12) Сполука 12 була одержана за схемою АЕ. Інтермедіат АЕ1 синтезували за наступною методикою. 2-(4Піридин)етансульфонову кислоту (3,0г, 0,016моль) розчинили у водному розчині соляної кислоти (2,0N, 12мл) та цей розчин обробили оксидом платини (IV) (0,13г). Цю суміш гідрогенезували при 50атм. та кімнатній температурі на протязі 21год., потім профільтрували через целіт та суміш випаровували, що дало гідрохлорид 2-(4-піперидил)етансульфонової кислоти у вигляді білого порошку (3,5г). Цей порошок розчинили у водному розчині тетрагідрофурану (1:1, 70мл) при кімнатній температурі обробили N-метилморфоліном (3,7мл, 2,2 еквіваленти) та бензилхлорформіатом (2,2мл, 1 еквіваленти). Цю суміш перемішували на протязі 15год., потім підкислили водним розчином лимонної кислоти та екстрагували хлороформом (двічі по 100мл). Органічну фазу висушили безводним сульфатом натрію та випарували, що дало 2-(4-Nбензилоксикарбонілпіперидин)етансульфонову кислоту (2,75г, у вигляді золотистої олії). Цю олію перетворили у кінцевий продукт 12 за п'ять послідовних стадій синтезу (Схема АЕ, W.J.Hoekstra, J.Med. Chem. 1995, 38, 1582) та отримали у вигляді безбарвної склоподібної речовини (0,060г): 1Н-ЯМР (ДМСО-D6) d 8,9 (м., 1H), 8,6 (м., 1H), 3,5 (м., 2Н), 3,1-3,3 (м., 4Н), 3,0 (м., 2Н), 2,6-2,8 (м., 4Н), 2,3 (м., 3Н), 1,65-1,9 (м., 5Н), 1,6 (м., 3Н); 1,21,4 (м., 5Н); мас-спектр: m/е 376 (МH+). Приклад 13 N-3-(4-піперидинпропіоніл)ніпекотил-5Н-(2-аміноетил)тетразола гідрохлорид (13) Сполука 13 була одержана за схемою АС. Інтермедіат АСІ (синтезований за методикою, описаною у W.J.Hoekstra, J.Med. Chem. 1995, 38, 1582) (1,9мілімоль) розчинили у дихлорметані (50мл) та обробили біс(2оксо-3-оксазолідиніл)фосфініл хлоридом (1,9мілімоль), N-метилморфоліном (1,9мілімоль) та 3амінопропіонітрилом (1,9мілімоль). Реакційну суміш струшували на протязі 18год., потім розбавили насиченим водним розчином хлориду амонію та розділили органічну та водну фази. Органічну фазу випаровували та отриманий продукт очистили хроматографією на силікагелі (10% етиловий спирт/дихлорметан), що дало олію. Олію розчинили у толуені (10мл), обробили азидотриметилсиланом (2,4мілімоль) та оксидом дибутилстануму (1,2мілімоль) та нагрівали із оберненим холоджильником на протязі 16год. Охолодження реакційної суміші дало осад брунатного кольору, який обробили діетиловим етером. Цю тверду речовину гідрогенізували над діоксидом платини (0,08г) у метанолі (12мл) та тиску 50атм. на протязі 15год., реакційну суміш профільтрували та розчинник випарували, що дало сполуку 13 у вигляді жовтої піни (0,065г): 1Н-ЯМР (ДМСОD6) d 8,9 (м., 1Н), 8,6 (м., 1H), 8,13 (д., J= 28, 1Н), 4,2 (м., 2Н), 3,2 (м., 3Н), 3,0 (м., 4Н), 2,7 (м., 4Н), 2,31 (к, J= 8, 2Н), 1,7-1,9 (м, 3Н), 1,4-1,6 (м, 5Н), 1,1-1,3 (м, 4Н); мас-спектр: m/е 364 (МH+). Приклад 14 N-3-(4-N-метилпіперазинпропіоніл)ніпекотил[3-аміно-3-(3,4-метилендіоксифеніл)]пропіонової кислоти натрієва сіль (14) Сполука 14 була одержана за схемою АВ. Етилніпекотат (3мілімоль) розчинили у дихлорметані (50мл), обробили акрилоілхлоридом (3мілімоль) та N-метилморфоліном (3мілімоль) та перемішували на протязі 1год. Розчинник випарували, залишок розчинили в етиловому спирті (50мл) та обробили N-метилпіперазином (3мілімоль). Розчин нагрівали при 60°С на протязі 15год., охолодили до кімнатної температури та розчинник випарували. Залишок розподілили між дихлорметаном (100мл) то водною фазою (10мл) та фази розділили. Органічну фазу висушили та випарували, що дало речовину у вигляді піни. Цю піну розчинили у воді, обробили водним розчином гідроксиду натрію (3мілімоль) та перемішували на протязі 1год., та випарували розчинник., що дало AB3·Na. Синтез закінчили за методикою, яка описана у W.J.Hoekstra, J.Месі. Chem. 1995, 38, 1582, використовуя метил 3-аміно-3-(3,4-метилендіоксифеніл)пропіонат (2,5мілімоль), що дало сполуку 14 у вигляді білої аморфної твердої речовини (0,14г): 1Н-ЯМР (D2O) d 6,8 (м., 3Н), 5,91 (с., 2Н), 5,0 (м., 1H), 4,0 (м., 1H), 3,7 (м., 1H), 2,8-3,4 (м., 11Н), 2,69 (с., 3Н), 2,4-2,6 (м., 7Н), 1,9 (м., 1Н), 1,7 (м., 2Н), 1,5 (м., 1H); мас-спектр: m/е 475 (МH+). Дані елементного аналізу для С 24H33N4O 6·Na·Н2 О (514,56). Розраховано: С, 56,02; Н, 6,86; N, 10,89. Знайдено: С, 55,72; Н, 6,78; N, 10,52. Приклад 15 N-3-(4-N-метилпіперазинпропіоніл)ніпекотил[3-аміно-3-(3-хінолініл)]пропіонової кислоти трис трифторацетат (15) Сполука 15 була одержана за методикою, яка описана у Прикладі 14. Синтез закінчили за методикою, яка описана у W.J.Hoekstra, J.Med. Chem. 1995, 38, 1582, використовуя метил 3-аміно-3-(3-хінолініл)пропіонат (6мілімоль) та АВ3 у якості реагентів. Сполуку 15 виділили у вигляді жовтого порошку (1,89г): 1Н-ЯМР (ДМСОD6) d 8,94 (c., 1H), 8,12 (с., 1H), 7,9 (м., 2Н), 7,6 (м., 2Н), 7,07 (д., J= 4, 1H), 5,2 (м., 1H), 4,1 (м., 1H), 3,7 (м., 1H), 3,1-3,3 (м., 2Н), 2,9 (м., 2Н), 2,6 (м., 2Н), 2,43 (с, ЗН), 1,9-2,4 (м., 12Н), 1,2-1,5 (м., 4Н); мас-спектр: m/е 482 (МН+). Приклад 16 N-3-(4-піперидинпрошоніл)-R-(-)-ніпекотил[(S)-3-аміно-3-(3,4-метилендіоксифеніл)]пропіонової кислоти гідрохлорид (16) До охолодженого розчину (5°С) t-бутоксикарбоніл-R-ніпекотової кислоти (9мілімоль) та метил (S)-3-аміно3-(3,4-метилендіоксифеніл)]пропіонату (див. Приклад із AG5) (9мілімоль) у ацетонітрилі (100мл) додали 2-(1Н бензотріазол-1-іл)-1,1,3,3-тетраметилсечовини гексафторфосфат (9мілімоль), гідроксибензотріазол (9мілімоль) та N-метилморфолін (18мілімоль). Цю суміш перемішували на протязі 15год., розбавили водою (10мл) та випарували. Залишок розбавили етилацетатом (100мл), органічну фазу висушили та потім випарували розчинник, що дало речовину у вигляді білої піни. Цю піну обробили розчином соляної кислоти (2N розчин у діоксані, 20мл), перемішували на протязі 3год. та випарували розчинник., що дало знову ж таки піну. Цю піну розчинили у ацетонітрилі (100мл) та обробили t-бутоксикарбонілпіперидинпропіоновою кислотою (7мілімоль), 2-(1H-бензотріазол-1-іл)-1,1,3,3-тетраметилсечовини гексафторфосфат (7мілімоль), гідроксибензотріазол (7мілімоль) та N-метилморфолін (14мілімоль). Цю суміш перемішували на протязі 6год., розбавили водою (10мл) та випарували. Залишок розбавили етилацетатом (100мл). Органічну фазу висушили, випарували та очистили хроматографією на силікагелі (7% етиловий спирт/дихлорметан), що дало речовину у вигляді піни. До розчину цієї піни (4,6мілімоль) у тетрагідрофурані при охолодженні на льодяній бані додали по краплинам LiOH·H2O (6,9мілімоль розчинили у 30мл води). Суміш перемішували на протязі 1,5год., підкислили оцтовою кислотою (1,7мл), та підігріли до кімнатної температури. Цей розчин розбавили хлороформом (75мл) та відділили органічну та водн у фази. Органічну фазу висушили безводним сульфатом натрію та випарували розчинник., що дало речовину у вигляді білої піни. Цю піну розчинили у суміші діоксану (20мл) та анізолу (0,3мл), охолодили на льодяній бані, обробили розчином соляної кислоти (15мл, 4,0N розчин у діоксані) та перемішували на протязі 3год., при цьому утворився осад речовини. Цей осад відфільтрували та промили діетиловим етером (150мл) та ацетонітрилом (20мл), що дало сполуку 16 у вигляді білого порошку (1,78г): Тпл. 190-200°С; 1Н-ЯМР (ДМСО-D6) d 8,9 (м., 1Н), 8,6 (м., 1Н), 8,4 (м., 1Н), 6,83 (д., J= 5, 1Н), 6,79 (д., J= 5, 1H), 6,7 (м., 1H), 5,95 (с., 2H), 5,08 (д.д., J= 5,11; 1Н), 4,1-4,3 (м., 1H), 3,7 (м., 1H), 3,15 (д., J= 10, 2Н), 3,0 (м., 1H), 2,7 (м., 2Н), 2,6 (м., 3Н), 2,31 (д., J= 7, 2Н), 1,81 (д., J= 10, 2Н), 1,2-1,7 (м., 11Н); мас-спектр: m/e 460 (МH+); [a]24D -0,478°(с 1,00, MeOH). Приклад 17 N-3-(4-піперидинпропіоніл)гексагідроазепін-3-карбоніл[3-аміно-3-(3-хіноліл)]пропіонової кислоти біс трифторацетат (17) Сполука 17 була одержана за схемою АА. Інтермедіат АА2 (0,36мілімоль) наситили 1,2-дихлоретаном (5мл) до набухання та обробили етил гексагідроазепін-3-карбоксилат гідрохлоридом (0,36мілімоль), діізопропілкарбодіімідом (0,72мілімоль) та діізопропілетиламіном (0,72мілімоль), потім струшували на протязі 16год. Розчинник видалили та смоляний сорбент промили (див. Приклад 1), а етиловий естер перетворили у відповідну кислоту взаємодією із калієм триметилсиланоатом (див. Приклад 1). Смоляний сорбент наситили диметилформамідом (5мл) до набухання та кислоту ввели в реакцію із метил 3-аміно-3-(3-хіноліл)пропіонатом (0,36мілімоль), та далі діяли за методикою, описаною в Прикладі 1. Сполука 17 була виділена у вигляді склоподібної речовини (0,10г): 1Н-ЯМР (D2O) d 9,06 (с, 1Н), 8,9 (м., 1H), 8,2 (м., 1H), 8,04 (с, 1H), 8,0 (т, J= 4, 2Н), 7,8 (т., J= 4, 2Н), 5,5 (м., 1H), 3,8 (м., 1H), 3,3 (м., 4Н), 3,0 (м., 2Н), 2,7 (м., 4Н), 2,0-2,4 (м., 6Н), 1,7-1,9 (м., 4Н), 1,1-1,6 (м., 8Н); мас-спектр: m/е 481 (МH+). Приклад 18 N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил[(S)-3-аміно-3-(3-хіноліл)]пропіонової кислоти біс гідрохлорид (18) Сполуку 18 синтезували за методикою, що описана у Прикладі 16, виходячі із t-бутоксикарбоніл-Rніпекотової кислоти (7,1мілімоль) та метил (S)-3-аміно-3-(3-хінолініл)пропіоната (див. Приклад із AG5) (7,1мілімоль). В результаті виділили речовину у вигляді лусок (1,11г): Тпл. 142-144°С; мас-спектру m/е 467 (МН+); [a] 24D -173° (с 0,1, МеОН). Дані елементного аналізу для С 26H34N4O4 ·2,25HCl·Н2 (566,64). Розраховано: С, 55,11; Н, 6,80; N, 9,89; СІ, 14,08. Знайдено: С, 54,85; Н, 6,62; N, 10,04; СІ, 13,68. Приклад 19 N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил[(S)-3-аміно-3-(2-t-бутилетиніл]пропіонової кислоти гідрохлорид (19) Сполуку 19 синтезували за методикою, що описана у Прикладі 16, виходячі із t-бутоксикарбоніл-Rніпекотової кислоти (7,1мілімоль) та метил (S)-3-aміно-3-(2-t-бутилетиніл)пропіоната (див. W.J.Hoekstra, J.Med. Chem. 1995, 38, 2378) (3,2мілімоль). В результаті виділили речовину у вигляді білого порошку (0,33г.): масспектр: m/е 420(МH+). Дані елементного аналізу для C23H37N 3O4 ·1,07HCI·0,43H2O (468,97). Розраховано: С, 59,21; Н, 8,42; N, 8,96; СІ, 8,09. Знайдено: С, 58,92; Н, 8,58; N, 8,76; СІ, 7,82. Приклад 20 N-3-(4-пшеридинпропіоніл)ніпекотил [(S)-3-аміно-3-(3,4-метилендіоксифеніл)]пропіонової кислоти біс трифторацетат (20) Сполука 20 була одержана за схемою AF. Інтермедіат AF3 (2,8мілімоль) розчинили у бензені (50мл), обробили етил ніпекотатом (2,8мілімоль) та реакційну суміш нагрівали на протязі 7год. Реакційну суміш охолодили, розподілили між водою (15мл) та етилацетатом (70мл) та розділили органічну та водну фази. Органічну фазу висушили та випарували, що дало AF4. AF4 перетворили в сполуку 20 за вже відомою методикою (W.J.Hoekstra, J.Med. Chem. 1995, 38, 1582), яку одержали у вигляді білого порошку (0,33г): 1HЯМР (CD3OD) d 8,6-8,8 (м., 3Н), 6,7-6,9 (м., 3Н), 5,91 (с, 2Н), 5,1-5,2 (м., 1Н), 3,3-3,5 (м., 4Н), 2,8-3,1 (м., 6Н), 2,6-2,7 (м., 3Н), 1,5-2,0 (м., 11H), 1,2-1,4 (м, 4Н); мас-спектр: m/е 446 (МH+). Приклад 21 N-3-(4-піперидинпропіоніл)-R-(-)ніпекотил[(S)-3-аміно-3-(3-піридил)]пропіонової кислоти біс трифторацетат (21) Сполуку 21 синтезували за методикою, що описана у Прикладі 16, виходячі із t-бутоксикарбоніл-Rніпекотової кислоти (6,4мілімоль) та метил (S)-3-аміно-3-(3-піридил)пропіоната (див. Приклад із AG5) (6,4мілімоль). В результаті виділили речовину у вигляді білої аморфної твердої речовини (1,60г): Тпл. 74-81°С; мас-спектр: m/е 417 (МH+). Дані елементного аналізу для C22H32N4O4·2,1C2HF3O 2·0,7Н2О (668,58). Розраховано: С, 47,07; Н, 5,35; N, 8,38; F, 17,90; KF, 1,89. Знайдено: С, 47,08; Н, 5,31; N, 8,41; F, 17,68; KF, 2,00. Приклад 22 N-3-(4-піперидинпропіоныл)-R-(-)-ніпекотил[(S)-2-(3-метоксіаніліно)карбоніламіно-3-аміноіпропіонова кислота (22) Метил t-бутоксикарбоніл-R-ніпекотил[(S)-2-бензилоксикарбоніламіно-3-аміно]пропіонат (9,5мілімоль) (синтезований із метил N-a-бензилоксикарбоніл-L-діамінопропіонату та t-бутоксикарбоніл-R-ніпекотової кислоти за методикою, описаною у Прикладі 16) розчинили у метиловому спирті (40мл) та гідрогенезували при тиску 50атм. над гідроксидом паладію (0,4г) на протязі 24год. Потім реакційну суміш профільтрували та розчинник випаровували, що дало АН2 у вигляді білої твердої речовини. АН2 (9,1мілімоль) розчинили у дихлорметані (100мл), охолодили (5°С), обробили 3-метоксифенілізоціанатом (9,1мілімоль) та Nметилморфоліном (9,1мілімоль) та перемішували на протязі 17год. Розчин розбавили насиченим водним розчином хлориду амонію (10мл), відділили органічну та водну фази, органічну фаз у висушили, випарували, що дало олію, яку очистили хроматографією на силікагелі (4% етиловий спирт/дихлорметан) та отримали сполуку АН3. Ін термедіат АН3 перетворили на сполуку 22 за 4 послідовні стадії синтезу, які описані у Прикладі 16, що дало аморфну тверду речовину білого кольору. (1,35г): Тпл. 72-76°С; 1Н-ЯМР (ДМСО-D6) d 8,7 (м., 3Н), 7,8 (м., 1H), 7,1 (м., 2Н), 6,8 (д., 1Н), 6,5 (д., 2Н), 3,66 (с., 3Н), 3,4 (м., 2Н), 3,2 (д., 2Н), 3,0 (м., 1H), 2,7 (д.д., 4Н), 2,3 (м., 3Н), 1,6 (м., 3Н), 1,1-1,7 (м., 11Н); мас-спектр: m/е 504 (МН+). Дані елементного аналізу для C25H37N5 O6 ·1,2HCl·1,0H2O (565,37). Розраховано: С, 53,11; Н, 7,17; N, 12,39; СІ, 7,53. Знайдено: С, 53,40; Н, 7,44; N, 12,14; СІ, 7,66. Використовуя подібну загальну методику, що описана у Прикладі 22, були синтезовані сполуки у Прикладах 26, 28-30 у відповідності зі схемою АН. У випадку карбаматних похідних у якості ацилюючого агента були використані відповідні алкілхлорформіати (аналогічно до перетворення АН2 у АН3; один мольний еквівалент). У випадку сульфонамідів в якості сульфонуючого агенту були використані відповідні сульфонілхлориди (один мольний еквівалент). Приклад 23 N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил[(S)-2-бензилоксикарбоніламіно-3-аміно]пропіонової кислоти гідрохлорид (23) Сполуку 23 синтезували за методикою, що описана у Прикладі 16, виходячі із t-бутоксикарбоніл-Rніпекотової кислоти (8,8мілімоль) та метил N-a-бензилоксикарбоніл-L-діамінопропіонату (8,8мілімоль). В результаті виділили речовину у вигляді білого порошку (1,65г): Тпл, 110-113°С; мас-спектр: m/е 489 (МH+). Дані елементного аналізу для С 25Н36Н4О 6·1,15НСІ·0,5Н2О·0,5діоксан (583,57). Розраховано: С, 55,56; Н, 7,41; N, 9,60; СІ, 6,99. Знайдено: С, 55,23; Н, 7,79; N,9,85; СІ, 7,01. Приклад 24 N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил[(S)-2-(3-хлорбензилокси)карбоніламіно-3-аміно]пропіонової кислоти гідрохлорид (24) Сполуку 24 синтезували за методикою, що описана у Прикладі 22, виходячі із 3хлорбензилоксикарбонілхлориду (6,6мілімоль) та АН2 (6,6мілімоль). В результаті виділили продукт у вигляді аморфної твердої речовини білого кольору (1,33г): T пл. 89-96°С; мас-спектр: m/e 524 (МH+). Дані елементного аналізу для С 25Н35СІN4О6 ·1,25НСІ·0,5Н2О·1,0дюксан (637,20). Розраховано: С, 50,89; Н, 7,08; N, 8,78; СІ, 12,52. Знайдено: С, 51,10; Н, 6,71; М, 8,38; СІ, 12,20. Приклад 25 N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил[(S)-2-бензилсульфоніламіно-3-аміно]пропіонової кислоти гідрохлорид (25) Сполуку 25 синтезували за методикою, що описана у Прикладі 22, виходячі із бензилсульфонілхлориду (5,2мілімоль) та АН2 (5,2мілімоль). В результаті виділили продукт у вигляді білого порошку (0,87г): Тпл. 145149°С; мас-спектр: m/e 509 (МH+). Дані елементного аналізу для C24H36N4O6S·1,3HCI·0,3 діоксан (568,06). Розраховано: С, 50,75; Н, 7,04; N, 9,86; СІ, 8,11. Знайдено: С, 51,03; Н, 6,93; N, 9,46; СІ, 7,85. Приклад 26 N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил[(S)-2-(3,5-диметоксіаніліно)карбоніламіно-3-аміно]пропіонової кислоти гідрохлорид (26) Сполуку 26 синтезували за методикою, що описана у Прикладі 22, виходячі із 3,5диметоксифенілізоціанату (10,2мілімоль) та АН2 (10,2мілімоль). В результаті виділили продукт у вигляді порошку білого кольору (1,89г.): Тпл, 190-193°С; мас-спектр: m/e 534 (МН+). Дані елементного аналізу для С26Н39N5 О7 ·1,2НСІ·0,2діоксан (585,40). Розраховано: С, 53,35; Н, 7,20; N, 11,96; СІ, 7,27. Знайдено: С, 53,48; Н, 7,38; N,12,05; СІ, 6,97. Приклад 27 N-[(4,4'-біпіперидин-1-іл)карбоніл]-R-(-)-ніпекотил[(S)-3-аміно-3-(3-піридил)]пропіонової кислоти трис гідрохлорид (27) Інтермедіат AJ1 (5,5мілімоль), який одержали за методикою, описаною в Прикладі 16, розчинили у дихлорметані (140мл), охолодили розчин (5°С), обробили р-нітрофенілхлороформіатом (5,5мілімоль) та Nметилморфоліном (16,5мілімоль) та перемішували на протязі 2 год. Суміш розбавили водою (15мл), відділили органічну та водну фази та органічну фазу висушили, й випарували дo стану олії. Цю олію розчинили в ацетонітрилі (70мл), обробили N-t-бутоксикарбаніл-4,4'-біпіперидином (7,5мілімоль) та 4диметиламінопіридином (5,5мілімоль) та нагрівали суміш із оберненим холодильником на протязі 24год. Реакційну суміш охолодили, випарували, що дало тверду речовину, яку очистили хроматографією на силікагелі (8% етиловий спирт/дихлорметан). В результаті отримали AJ2 у вигляді склоподібної речовини зеленого кольору (1,5моль). AJ2 омилили та провели зняття захисту за методикою, описаною у Прикладі 16, що дало сполуку 27 у вигляді рожево-жовтого порошку (0,73г): Тпл. 121-125°С; мас-спектр: m/е 472 (МН+). Дані елементного аналізу для С 25Н 37N5 О4 ·3,6НСІ·1,0діоксан (690,98). Розраховано: С, 50,41; Н, 7,09; N, 10,14; СІ, 18,47. Знайдено: С, 50,80; Н, 7,31; N, 10,20; СІ, 18,78. Приклад 28 N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил[(S)-2-(2-нафтиламіно)карбонІламіно-3-аміноіпропіонової кислоти гідрохлорид (28) Сполуку 28 синтезували за методикою, що описана у Прикладі 22, виходячі із 2-нафтилізоціанату (8,5мілімоль) та АН2 (8,5мілімоль). В результаті виділили продукт у вигляді білого порошку (1,65г.): Тпл. 187193°С; мас-спектр: m/е 524 (МН+). Дані елементного аналізу для С 28Н37Н5 О5 ·1,36НСІ·0,72діоксан (602,07). Розраховано: С, 55,86; Н, 7,39; N, 11,63; СІ, 8,01. Знайдено: С, 56,03; Н, 7,11; N, 11,23; СІ, 7,97. Приклад 29 N-3-(4-піперидинпропюніл)-R-(-)-ніпекотиламінометил-5-(S)-(3-N-бензил)імідазолін 2,4-діона гідрохлорид (29) N-3-(4-піперидинпропіоніл)-R-(-)-ніпекотил[(S)-2-(2-бензиламіно)карбоніламіно-3-аміно]пропіонової кислоти гідрохлорид (0,15г), який синтезували виходячі із інтермедіата АН2 (4,4мілімоль) та бензилізоціанату (4,4мілімоль) за методикою, що описана у Прикладі 22, розчинили у водному розчині соляної кислоти (3N) та перемішували на протязі 18год. при кімнатній температурі. Цей розчин сконцентрували у вакуумі, що дало тверду речовину білого кольору. Цю тверду речовину розтерли у порошок та висушили, що дало сполуку 29 у вигляді білої піни (0,144г.): 1Н-ЯМР (ДМСО-D6) d 9,0 (м., 1Н), 8,6 (м., 1H), 8,3 (м., 1H), 7,2 (м., 5Н), 4,48 (с, 2Н), 4,2 (м., 2Н), 3,7 (м., 1H), 3,4 (м., 1H), 3,2 (д., 3Н), 2,7 (д., 3Н), 2,2 (м., 3Н), 1,7 (м., 3Н), 1,0-1,6 (м., 10Н); масспектр: m/е 470 (МH+). Приклад 30 N-3-(4-піперидинпропюніл)-R-(-)-ніпекотил[(S)-2-(2-фенетиламіно)карбоніламіно-3-аміно]пропіонової кислоти форміат (30) Сполуку 30 синтезували за методикою, що описана у Прикладі 22, виходячі із 2-фенетилізоціанату (4,1мілімоль) та АН2 (4,1мілімоль). В результаті виділили продукт у вигляді піни жовто-брунатного кольору (0,41г): Тпл. 65-72°С; мас-спектр: т/е 502 (МH+). Дані елементного аналізу для С 26Н39Н5О5 ·1,2НСО2Н·1,0Н2О (574,87). Розраховано: С, 56,83; Н, 7,61; N, 12,18. Знайдено: С, 57,12; Н, 7,80; N, 11,85. 6-Метил-3-піридинкарбоксальдегід (АК2) Прекурсор АК2, який є альдегідом, одержали в дві стадії, використовуя загальноприйнятій методи синтезу. АК1 (0,066мілімоль) розчинили у тетрагідрофурані (100мл), розчин охолодили (-78°С), обробили літій алюмогідридом (LiAIH4) (0,066мілімоль) та перемішували реакційну суміш на протязі 4год. Потім до суміші додали насичений водний розчин хлориду амонію, підігріли, відфільтрували, промили хлороформом (250мл) та відділили водну й органічну фази. Органічну фазу висушили та випарували, що дало безбарвну олію (0,054мілімоль). Цю олію розчинили у дихлорметані (200мл), обробили діоксидом мангану (МnО2) (70г) та нагрівали із оберненим холодильником на протязі 6год. Потім суміш охолодили, відфільтрували та розчинник випарували, що дало сполуку АК2 (0,052мілімоль) у вигляді брунатної олії. Приклад 31 N-3-(4-піперидинпропіоніл)-R-(-)ніпекотил[(S)-3-аміно-3-(6-метил-3-піридил)]пропіонової кислоти біс гідрохлорид (31) Сполуку 31 синтезували за методикою, що описана у Прикладі 16, виходячі із t-бутоксикарбоніл-Rніпекотової кислоти (6,9мілімоль) та метил (S)-3-аміно-3-(6-метил-3-піридил)]пропіонату (6,9мілімоль) (див. Приклади із АК5 та AG5). В результаті виділили сполуку 31 у вигляді білої піни (1,20г): Тпл. 99-105°С; масспектр: m/е 431 (МH+). Дані елементного аналізу для С 23Н34N4О 4·2,24НСІ·1,0Н2О·0,24ацетонітрил (534,33). Розраховано: С, 51,70; Н, 7,35;N, 11,11; СІ, 14,82. Знайдено: С, 51,32; Н, 7,45; N, 11,23; СІ, 14,42. Приклад 32 N-3-(4-піперидинпропіоніл)-R-(-)ніпекотил[(S)-3-аміно-3-(5-бромо-3-піридил)]пропіонової кислоти біс гідрохлорид (32) Сполуку 32 синтезували за методикою, що описана у Прикладі 16, виходячі із t-бутоксикарбоніл-Rніпекотової кислоти (4,8мілімоль) та метил (S)-3-аміно-3-(5-бромо-3-піридил)]пропіонату (4,8мілімоль) (див. Приклади із АК5 та AG5). В результаті виділили сполуку 32 у вигляді білої піни (1,24г): Тпл. 98-101°С; масспектр: m/е 496 (МH+). Дані елементного аналізу для C22H31BrN4O 4·2,2HCI·1,0H2O (593,67). Розраховано: С, 44,51; Н, 5,98; N, 9,44; СІ, 13,14. Знайдено: С, 44,17; Н, 6,37; N,9,81; СІ, 13,10. Приклад 33 N-3-(4-формамідинопіперидинпропіоніл)-R-(-)-ніпекотил[(S)-3-аміно-3-(3-піридил)]пропіонової кислоти біс гідрохлорид (33) Сполуку 33 синтезували за методикою, що описана у М.К.Scott (J.Med. Chem., 1983, 26, 534), як показано на схемі AL. Інтермедіат AL1 (див. Приклад 21) (2,3мілімоль) розчинили в етиловому спирті (20мл), обробили етилформімідат гідрохлоридом (3,7мілімоль), перемішували на протязі 22год. та відфільтрували. Фільтрат обробили діетиловим етером (40мл), охолодили на льодяній бані та осад, що випав відфільтрували. В результаті виділили склоподібний продукт AL2, який розчинили у водному розчині соляної кислоти (4N, 15мл), перемішували на протязі 28год. та потім випарували розчинник, що дало сполуку 33 у вигляді білої піни (0,75г): Тпл. 49-55°С; 1Н-ЯМР (ДМС О-D6) d 9,35 (а, 1H), 9,1 (м., 2Н), 8,8 (м., 2Н), 8,70 (д., 1H), 8,5 (м., 1H), 7,8 (м., 2Н), 5,2 (д.д., 1H), 4,2 (м., 1Н), 3,8 (м., 2Н), 3,2 (м., 2Н), 2,8 (м., 2Н), 2,6 (м., 1H), 2,3 (м., 2Н), 1,8 (м., 3Н), 1,0-1,7 (м., 12Н); мас-спектр: m/е 444 (МH+).
ДивитисяДодаткова інформація
Назва патенту англійськоюCarboxamide derivatives of piperidine, a composition based thereon and a method for treatment of thrombotic disorders
Назва патенту російськоюКарбоксамидные производные пиперидина, композиция на их основе и способ лечения тромбоцитарных расстройств
МПК / Мітки
МПК: C07D 211/60, C07D 405/12, A61K 31/472, A61K 31/55, A61K 31/4427, A61K 31/47, A61K 31/4406, C07D 401/14, A61P 7/00, A61K 31/495, C07F 7/08, A61K 31/00, A61K 31/443, C07D 213/55, A61P 7/02, A61K 31/4545, C07D 401/12, A61K 31/4465, C12P 17/10, A61K 31/445, A61K 31/4523, C07D 405/14, A61K 31/44, A61K 31/4725, C07D 401/06
Мітки: спосіб, композиція, карбоксамідні, похідні, розладів, тромбоцитарних, лікування, піперидину, основі
Код посилання
<a href="https://ua.patents.su/17-70285-karboksamidni-pokhidni-piperidinu-kompoziciya-na-kh-osnovi-ta-sposib-likuvannya-trombocitarnikh-rozladiv.html" target="_blank" rel="follow" title="База патентів України">Карбоксамідні похідні піперидину, композиція на їх основі та спосіб лікування тромбоцитарних розладів</a>
Похідні піперазину та піперидину, спосіб їх отримання, фармацевтична композиція, спосіб отримання фармацевтичної композиції і спосіб лікування розладів цнс

Номер патенту: 52656
Опубліковано: 15.01.2003
Автори: Лонг Стівен К., Тульп Мартінус Т.М., Крузе Конеліс Г., Фенстра Рулоф В., КЬОЙПЕРС Вільма
МПК: C07D 407/10, C07D 209/08, C07D 413/12, C07D 215/38, C07D 215/12, C07D 409/12, C07D 319/00, C07D 265/36, C07D 307/79, A61K 31/55, A61K 31/34, A61K 31/551, A61K 31/496, C07D 409/10, A61K 31/495, A61K 31/357, C07D 407/12, C07D 413/10, C07D 321/00, C07D 317/66, A61K 31/00, A61P 43/00, A61K 31/42, C07D 405/04, C07D 333/54, A61K 31/5513, A61K 31/423, C07D 311/18, C07D 327/00, A61K 31/343, C07D 263/58, A61P 25/04
Мітки: композиція, похідні, піперидину, композиції, фармацевтично, цнс, фармацевтична, піперазину, розладів, отримання, лікування, спосіб
Формула / Реферат:
1. Сполуки загальної формули (а),,де- А являє собою гетероциклічну групу з 5-7 кільцевих атомів, у якій присутні 1-3 гетероатоми з групи О, N та S,- R1 - водень чи фтор ,- R2 - С1-4 - алкіл, С1-4-алкокси або оксогрупа, а р дорівнює 0, 1 чи 2,- Z являє собою вуглець чи азот, а пунктирною лінією позначено простий зв'язок, якщо Z - азот або простий чи подвійний зв'язок, якщо Z - вуглець,- R3 та R4...
Похідні піперидину з аналгезивною дією, спосіб їх отримання та фармацевтична композиція на їх основі

Номер патенту: 65552
Опубліковано: 15.04.2004
Автори: Робертс Едвард, Вей Жонгйонг, Делорм Даніель
МПК: C07D 409/06, A61K 31/4427, C07D 211/70, C07F 9/59, C07D 405/06, A61K 31/4409, C07D 401/06, A61K 31/661, C07D 211/22, A61K 31/4709, A61K 31/4436, A61P 29/00, C07D 211/46, A61K 31/443
Мітки: спосіб, похідні, отримання, фармацевтична, аналгезивною, основі, піперидину, дією, композиція
Формула / Реферат:
1. Сполука загальної формули (І),(I)де R1 вибирають з водню, розгалуженого або нерозгалуженого С1-С6-алкілу, С1-С6-алкенілу, С3-С8-циклоалкілу, С4-С8-(алкілциклоалкілу), де алкіл являє собою С1-С2-алкіл, і циклоалкіл являє собою С3-С6-циклоалкіл;С6-С10-арилу або гетероарилу, що має від 5 до 10 атомів, вибраних з С, S, N і О; де арил або гетероарил може бути необов'язково і незалежно заміщений 1 або 2 замісниками,...
Фторалкоксифенільні похідні піперидину або хінуклідину, що є антагоністами речовини p і фармацевтична композиція на їх основі

Номер патенту: 39168
Опубліковано: 15.06.2001
Автори: Лоу Джон Адамс ІІІ, РОУЗЕН Террі Джей
МПК: C07D 487/08, A61P 29/00, C07D 471/18, C07D 211/76, A61P 25/26, C07D 215/42, A61P 25/24, C07D 471/08, A61P 25/20, C07D 453/00, A61K 31/445, C07C 45/44, C07C 45/67, A61K 31/55, A61P 37/08, A61P 25/18, A61K 31/47, C07D 211/56, A61K 31/473, C07D 498/18, A61K 31/451, C07C 45/51, A61K 31/435, C07C 45/71, C07C 47/52, C07D 209/52, C07D 513/18, C07D 207/14, C07D 221/04
Мітки: речовини, фторалкоксифенільні, композиція, основі, фармацевтична, хінуклідину, піперидину, антагоністами, похідні
Формула / Реферат:
1. Фторалкоксифенильные производные пиперидина и хинуклидина формулы (I): , (I)где X1 представляет собой водород, С1-С3 алкокси, необязательно замещенную двумя-тремя атомами фтора, или С1-С4 алкил;X2 и X3 независимо выбирают из водорода, галогена, нитро, С1-С4...
Похідні амідів гетероарил-гексанових кислот, фармацевтична композиція для лікування чи попередження розладів чи станів (варіанти) та спосіб лікування чи попередження розладів чи станів (варіанти)

Номер патенту: 67735
Опубліковано: 15.07.2004
Автори: Посс Крістофер Стенлі, Кет Джон Чарлз, Браун Метью Френк
МПК: A61K 31/4184, C07C 237/22, C07D 277/68, C07D 237/28, C07D 403/12, C07D 213/82, A61K 31/47, C07D 235/24, A61K 31/44, C07D 209/42, A61P 1/00, A61P 17/00, C07D 215/48, C07D 213/81, A61K 31/381, A61K 31/498, C07D 241/44, C07D 471/04, A61P 29/00, C07D 333/70, C07D 409/12, A61K 31/455, A61K 31/428, A61P 37/02, C07D 215/54, A61K 31/343, C07D 307/85, C07D 241/24, C07D 417/12, A61P 43/00, C07D 221/04, A61P 3/10, A61K 31/502, C07D 405/12, C07D 401/12, C07D 217/26, C07D 221/12, C07D 521/00, A61K 31/404
Мітки: композиція, розладів, похідні, спосіб, попередження, станів, фармацевтична, гетероарил-гексанових, варіанти, лікування, кислот, амідів
Формула / Реферат:
1. Похідні амідів гетероарил-гексанових кислот формули ,де R1 – (C2-C9)гетероарил, заміщений, як варіант, одним чи більше замісниками, незалежно вибраними з групи, яка складається з гідрогену, галогену, CN, (C1-C6)алкілу, заміщеного, як варіант, одним чи більше атомами флуору, гідроксилу, гідроксі(C1-C6)алкілу, (C1-C6)алкоксилу, заміщеного, як варіант, одним чи більше атомами флуору, (C1-C6)алкоксі(C1-C6)алкілу, НО-(C=О)-,...
Заміщені 6,6-гетеробіциклічні похідні, проміжні сполуки, фармацевтична композиція та спосіб лікування, попередження або інгібування розладів

Номер патенту: 66345
Опубліковано: 17.05.2004
Автор: Чен Юпін Лян
МПК: C07D 475/00, A61P 25/24, A61K 31/47, C07D 215/22, A61K 31/00, A61P 5/24, A61P 25/30, A61K 31/4353, A61P 37/04, A61K 31/5365, A61K 31/529, A61P 1/00, A61P 31/00, A61P 37/00, A61P 15/00, A61P 5/42, A61K 31/519, A61P 21/00, A61P 25/20, A61P 25/22, A61K 31/4709, A61P 3/00, C07D 215/42, A61P 43/00, A61P 29/00, A61K 31/4985, A61P 3/02, A61P 1/04, C07D 215/233, A61P 13/00, A61P 13/02, A61P 25/16, A61P 1/14, C07D 498/04, A61P 25/28, A61K 31/495, A61P 15/08, A61P 25/04, A61K 31/535, A61K 31/4704, C07D 215/18, C07D 471/04, A61K 31/4375, A61K 31/505, A61K 31/435, A61P 35/00, A61P 31/18, A61P 3/04, A61P 1/12, A61P 15/06, C07D 405/12, A61P 21/02, A61P 25/08
Мітки: проміжні, фармацевтична, інгібування, розладів, попередження, сполуки, 6,6-гетеробіциклічні, похідні, композиція, заміщені, лікування, спосіб
Формула / Реферат:
(21) 99020647(57)1. Заміщені 6, 6-гетеробіциклічні похідні формули, Iде пунктирна лінія означає можливі подвійні зв'язки;А - нітроген чи CR7;В - -NR1R2, -CR1R2R10, -C(=CR2R11)R1, -NHCR1R2R10, -OCR1R2R10, -SCR1R2R10, -CR2R10NHR1, -CR2R10OR1, -CR2R10SR1 або -COR2;G - нітроген чи CR4, і з всіма оточуючими атомами пов'язаний...
Попередній патент: Спосіб виробництва натуральних фруктових і/або овочевих порошків
Випадковий патент: Захист стійки стелажа