Спосіб одержання лозартрану







Формула / Реферат
1. Спосіб одержання калієвої солі 2-бутил-4-хлор-1-[[(2'-1Н-тетразол-5-іл)-1,1'-біфеніл-4-іл]метил]-5-гідроксиметилімідазолу - лозартрану формули III
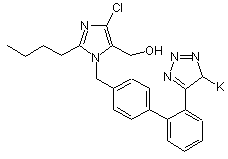 (III),
(III),
який відрізняється тим, що проводять сольволіз сполуки 2-бутил-4-хлор-1-[[2'-(1-тритил-1H-тетразол-5-іл)-1,1'-біфеніл-4-іл]метил]-5-гідроксиметилімідазол формули IV
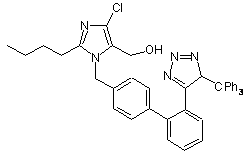 (IV)
(IV)
в безводному метанолі в нейтральному або слабоосновному середовищі і реакцію з еквівалентом речовини формули MnB, де М являє собою калій, n приймає значення 1 або 2 і B є CO32- або HCO32-.
2. Спосіб за п. 1, який відрізняється тим, що сольволіз проводять в слабоосновному середовищі і карбонат кальцію або гідрокарбонат калію додають на початку реакції.
3. Спосіб за пп. 1 або 2, який відрізняється тим, що продукт кристалізують з суміші спирту, переважно ізопропанолу, і антирозчинника, в якому сполука формули (ІІІ) є нерозчинною, або використовують інший розчинник, наприклад ацетон.
Текст
1. Спосіб одержання калієвої солі 2-бутил-4хлор-1-[[(2'-1Н-тетразол-5-іл)-1,1'-біфеніл-4іл]метил]-5-гідроксиметилімідазолу - лозартрану формули III Cl N N N N OH N N N CPh3 (IV) в безводному метанолі в нейтральному або слабоосновному середовищі і реакцію з еквівалентом речовини формули MnB, де М являє собою калій, n приймає значення 1 або 2 і B є CO32- або HCO32-. 2. Спосіб за п. 1, який відрізняється тим, що сольволіз проводять в слабоосновному середовищі і карбонат кальцію або гідрокарбонат калію додають на початку реакції. 3. Спосіб за пп. 1 або 2, який відрізняється тим, що продукт кристалізують з суміші спирту, переважно ізопропанолу, і антирозчинника, в якому сполука формули (ІІІ) є нерозчинною, або використовують інший розчинник, наприклад ацетон. 86947 N (13) (III), який відрізняється тим, що проводять сольволіз сполуки 2-бутил-4-хлор-1-[[2'-(1-тритил-1Hтетразол-5-іл)-1,1'-біфеніл-4-іл]метил]-5гідроксиметилімідазол формули IV Cl N C2 K (11) OH UA N (19) (21) a200603282 (22) 26.08.2004 (24) 10.06.2009 (86) PCT/CZ2004/000051, 26.08.2004 (31) PV 2003-2319 (32) 27.08.2003 (33) CZ (31) PV 2004-733 (32) 16.06.2004 (33) CZ (46) 10.06.2009, Бюл.№ 11, 2009 р. (72) РАДЛ СТАНІСЛАВ, CZ, СТАХ ЯН, CZ, КЛЕЦАН ОНДРЕЙ, CZ (73) ЗЕНТІВА А.С., CZ (56) EP, 0797121, A2, 24.09.1997 WO, 02094816, A1, 28.11.2002 US, 5559233, A, 24.09.1996 US, 5399578, A, 21.03.1995 US, 5196444, A, 23.03.1993 US, 5281604, A, 25.01.1994 NICHOLAS, J. S. HARMAT ET AL: "4-Diazinyl- and 4-Pyridinylimidazoles: Potent Angiotensin II Antagonists. A Study of Their Activity and Computational Characterization" JOURNAL OF MEDICINAL CHEMISTRY, vol. 38, no. 15, 1995, pages 2925-2937 CARINI D J ET AL: "NONPEPTIDE ANGIOTENSIN II RECEPTOR ANTAGONISTS: THE DISCOVERY OF A SERIES OF N(BIPHENYLYLMETHYL)IMIDAZOLES AS POTENT, ORALLY ACTIVE ANTIHYPERTENSIVES" JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 34, no. 8, 1 August 1991 (1991-08-01), pages 2525-2547 USIFOH, CYRIL O.: "Anticonvulsant activity of reaction products of 5,5-diphenylhydantoin with substituted methylene bromides" ARCH. PHARM. PHARM. MED. CHEM., vol. 334, no. 11, 2001, pages 366-368 ATTANASI O. A. ET AL.: "Synthesis of biphenyltetrazole derivatives of 1-aminopyrroles as angiotensin II antagonists" FARMACO, vol. 54, 1999, pages 64-76 2 3 Цей винахід стосується покращеного способу видалення трифенілметанової (тритильної) захисної групи з 1-трифенілметил-5-(4'-заміщений амінометил-1,1'-біфеніл-2-іл)-1Н-тетразолів загальної формули І в якій R означає наступні групи і де R1, R2 і R3 може бути Н, галогеном, нерозгалуженим або розгалуженим С1-С5 алкілом, С1-С5 гідроксиалкілом, С1-C5 алкокси, С1-С5 алкоксиметилом або бензилом, або R2 і R3 можуть разом утворювати С5-С7 насичене або ненасичене кільце, необов'язково незаміщене або заміщене ароматичне кільце, і способу його застосування для виробництва лікарського засобу для регулювання кров'яного тиску з групи антагоністів ангіотензину II загальної формули II в якій R може мати ті ж самі значення, що і в загальній формулі І, і в якій Μ є або воднем, або лужним металом. Калієву сіль лозартану формули III одержують згідно з опублікованими способами (WO 95/17396, ЕР 253310, US 5,859,258; J. Med. Chem. 1991, 34, 2525; J. Org. Chem. 1994, 59, 6391) за допомогою декількох методик, як використовують тритиллозартан формули IV як головну проміжну сполуку. Згідно з оригінальними патентами тритиллозартан формули IV був перетворений кислотним гідролізом у 2-бутил-4-хлор-1-[[(2'-тетразол-5-іл)1,1'-біфеніл-4-іл]метил]-5-гідроксиметилімідазол, який в подальшому згадується як "лозартанова кислота", формули V 86947 4 який було виділено і потім перетворено за допомогою гідроксиду калію в калієву сіль лозартану формули III. При видаленні тритильної захисної групи зазвичай використовують сильно корозивні кислоти. Необхідність виділення вільної кислоти лозартану та складне виділення надлишку мінеральних кислот з продукту є недоліками цього способу. Вільну кислоту, одержану в такий спосіб, потім перетворюють за допомогою водного гідроксиду калію в калієву сіль, яку потім, відповідно до вищезгаданих патентів, розчиняють в ізопропанолі, і продукт кристалізують після азеотропної перегонки з циклогексаном. Недоліком тут є особливо довготривала азеотропна перегонка. На основі більш нових патентних заявок (WO 01/61336; WO 02/094816) тритильна захисна група може також бути видалена дією сильно лужного розчину гідроксиду калію в первинних спиртах. Цим способом може бути отримана калієва сіль лозартану формули III, і наступну кристалізацію проводять додаванням розчинника, в якому калієва сіль лозартану є нерозчинною. Однак, в процесі згаданого лужного детритилювання сильною основою утворюються деякі незначні домішки, і видалити їх з продукту досить складно. Однією з найкращих можливостей синтезувати ірбезартан формули VI є синтез через тритилірбезартан формули VII описаний в патенті (US 5,559,233). Видаленням тритильної захисної групи отримують безпосередньо ірбезартан формули VI. Вищезгаданий патент також використовує детритилювання в кислому середовищі, яке має вже обговорювані недоліки. Головною проміжною сполукою одного з найбільш переважних способів синтезу валзартану формули VIII є бензиловий естер тритилвалзартану формули IX Валзартан формули VIII отримують згідно з опублікованим патентом (US 5,399,578) в такий 5 86947 6 спосіб, що бензиловий естер тритилвалзартану груп, крім того, застосування каталізатора, що місформули IX детритилюють дією соляної кислоти в тить паладій, збільшує витрати. В обох випадках діоксані, отримуючи, таким чином, бензиловий трифенілметанол або трифенілметан, які утворюестер валзартану формули X ються в процесі реакцій, повинні бути видалені складним екстрагуванням. Крім вищезгаданих способів детритилювання, для подібних речовин зартанового типу описують також детритилювання, каталізоване безводними кислотами в безводних спиртах, переважно в метанолі (US 5,763,619). Згідно з інформацією в згаНа другій стадії захисну групу бензилового есданому патенті перевага такого способу полягає в теру видаляють каталітичним гідруванням і отритому, що не відбувається відщеплення інших здамують валзартан формули VIII. тних до гідролізу функціональних груп. Інший спосіб використовують для міченого ізоКандезартран цилексетил одержують згідно з топами валзартану, де обидві захисні групи видаопублікованими патентами (US патент 5,196,444 і ляють каталітичним гідруванням (І. Labelled. Cpd. US патент 5,763,619), використовуючи наступний Radiopharm. 2000, 43, 1245). Недоліком першого спосіб: способу є використання корозивної соляної кислоти. При каталітичному гідруванні обох захисних Синтез починають з 1(циклогексилоксикарбонілокси)етил-2-етокси-1-[[2'(N-трифенілметил-1Н-тетразол-5-іл)біфеніл-4іл]метил]бензімідазо-7-карбоксилату формули XI, який в метанолі за допомогою соляної кислоти перетворюють в кандезартан цилексетил формули XII. Синтез вихідної речовини XI описано в основному патенті (US патент 5,196,444) і сполука в наш час є наявною у продажі. Спосіб детрити л ювання, описаний в основному патенті (US патент 5,196,444), має дуже низький вихід, і продукт повинен бути очищений за допомогою хроматографії. Фірма Takeda company покращила цю головну стадію шляхом використання безводного хлористого водню в метанолі (US патент 5,763,619), при цьому частина продуктів розкладання є нижчою, а вихід - вищим. В патенті US 5,763,619 цей метод не використовують для детритилювання жодної проміжної сполуки, придатної для одержання лозартану, ірбезартану або валзартану. Принаймні, у випадку валзартану ймовірно повинна відбуватись часткова переестерифікація і, можливо, часткове відщеплення валероїлового залишку. Аналогічно, у випадку ірбезартану можна також очікувати розкриття дигідроімідазолонового кільця. Іншими недоліками вважаються коливання виходів (в прикладах вони коливаються від 42% до 92%), агресивність реакційного середовища та необхідність використання води для видалення надлишків використовуваної кислоти, що частково нівелюють переваги реакції в безводному середовищі. Крім того, у випадку лікарських препаратів, використовуваних у формі солей лужних металів (наприклад, лозартан), необхідно потім перетворювати виділену "кислоту" у відповідну сіль. Приймаючи до уваги той факт, що найкращою використовуваною кислотою є розчин безводного хлористого водню в безводному спирті, необхідність одержання безводного розчину використовуваної кислоти у відповідному спирті є також важливим недоліком. Недоліки вищезгаданих способів включають застосування сильно корозійних кислот, а також необхідність обробляти реакційну суміш складними екстракціями. Таке виробництво є до того ж економічно невигідним. Об'єктом винаходу є покращений спосіб видалення трифенілметанової (тритильної) захисної групи з 1-трифенілметил-5-(4'-заміщений амінометил-1,1'-біфеніл-2-іл)-1Н-тетразолів і спосіб його використання для одержання калієвої солі 2бутил-4-хлор-1-[[(2'-1Н-тетразол-5-іл)-1,1'-біфеніл4-іл]метил]-5-гідроксиметилімідазолу (лозартану) формули III 2-бутил-3-[[2'-(1H-тетразол-5-іл)-1,1'-біфеніл-4іл]метил]-1,3-діазаспіро[4.4]нон-1-ен-4-ону (ірбезартану) формули VI N-(1-оксопентил)-N-[[2'-(1Н-тетразол-5-іл)-1,1'біфеніл-4-іл]метил-L-валіну (валзартан) формули VIII 7 86947 8 реважно при кип'ятінні зі зворотним холодильником, коли реакція завершується в межах декількох годин. Якщо зберігаються абсолютно безводні умови, в процесі реакції утворюється метилтрифенілметиловий етер формули XIII, який, після закінчення реакції, легко видаляється фільтрацією та 1-(циклогексилоксикарбонілокси)етил-2після охолодження метанольного розчину. Замість етокси-1-[[2'-(1Н-тетразол-5-іл)біфеніл-4метанолу можуть також використовуватися інші іл]метил]бензімідазол-7-карбоксилату (кандезарпервинні спирти, наприклад, етанол, але час реактан цилексетил) формули XII ції потому суттєво збільшується. Реакцію можна проводити також в суміші метанолу з іншими розчинниками, наприклад, з іншими спиртами, переважно, з етанолом, галогенованими розчинниками, переважно з дихлорметаном та хлороформом, Згадані лікарські препарати, які є терапевтичаліфатичними кетонами, переважно з ацетоном но важливими засобами, використовуваними для або 2-бутаноном, діалкіловими етерами, переважрегуляції кров'яного тиску, належать до медичної но з діізопропіловим етером та метил mpemгрупи так званих антагоністів рецепторів ангіотенбутиловим етером, та естерами карбонових кисзину II. лот з аліфатичними спиртами, переважно з меЦей спосіб заснований на несподіваному відтилацетатом, етилацетатом, ізопропілацетатом критті того, що видалення тритильної захисної або етилпропіонатом. В такому випадку, після тогрупи з 1-трифенілметил-5-(4'-заміщений аміномего, як реакція закінчується, суміш випарюють до тил-1,1'-біфеніл-2-іл)-1Н-тетразолів загальної фосухого залишку, потім розчиняють при високій термули І, особливо з тритильних похідних формул мпературі в метанолі і після охолодження суміші IV, VII і IX та XI, може бути здійснене шляхом сообробляють, як описано вище. льволізу при кип'ятінні зі зворотним холодильниЯкщо вихідною тритильованою проміжною 1 ком в безводному С -С5 спирті, переважно в безсполукою є тритиллозартан формули IV, згаданим водному метанолі або в суміші метанолу з способом одержують розчин вільної "лозартанової розчинником, що змішується з ним, без присутноскислоти" формули V і потім перетворюють в калієті будь-якого кислотного або основного агентів. ву сіль лозартану формули III дією карбонату ка"Лозартанову кислоту" формули V, отриману в лію, гідрокарбонату калію або гідроксиду калію. такий спосіб з тритиллозартану формули IV, потім Потім сама кристалізація може бути проведена із перетворюють під дією слабкої основи, наприклад, суміші спирту, переважно ізопропанолу, та нерозгідрокарбонату калію або карбонату калію, в калієчинника, в якому калієва сіль лозартану формули ву сіль лозартану формули III. Перетворення триIII є нерозчинною, або з використанням інших розтиллозартану формули IV в калієву сіль лозартану чинників, наприклад, ацетону. При використанні формули III може також бути здійснене шляхом цього способу може бути отриманий надзвичайно додавання згаданої слабкої основи на початку чистий продукт, що не містить домішок, які є звиреакції. чайними для кислотного способу або для способу З тритилірбезартану (VII) способом представз використанням гідроксиду калію. Зняття захисту леного винаходу безпосередньо утворюється ірбебез суттєвого погіршення чистоти неочищеної казартан (VI), який є достатньо чистим для того, щоб лієвої солі лозартану формули III також може бути бути придатним для використання як лікарської проведене безпосередньо в присутності слабкої речовини після простої кристалізації. основи, переважно карбонату або гідрокарбонату Бензиловий естер тритилвалзартану формули калію, при цьому безпосередньо продуктом є згаIX перетворюють згідно із способом представленодана калієва сіль лозартану формули III. го винаходу в бензиловий естер валзартану форЯкщо вихідною тритильованою проміжною мули X, який легко позбавляють надлишку утворесполукою є тритилірбезартан формули VII, згаданого метилтрифенілового етеру формули XIII ним способом одержують розчин ірбезартану формули VI; більшу частину метилтрифенілметилового етеру формули XIII видаляють шляхом концентрування та охолодження розчину. Високочистий ірбезартан може бути отриманий шляхом подальшого очищення кристалізацією з відповіді потому дебензилюють одним з описаних споних розчинників, наприклад, етанолу або ізопрособів до валзартану формули VIII. панолу. Кандезартан цилексетил, утворений описаним Якщо вихідною тритильованою проміжною детритилюванням, переважно може бути кристалісполукою є бензиловий естер тритилвалзартану зований з розчинників, в яких він легко розчиняформули IX, його, з використанням способу предється, або з розчинників, в яких він частково розставленого винаходу, перетворюють в бензиловий чинний. Особлива перевага надається естер валзартану формули X, який легко позбавкристалізації з їх суміші. ляють надлишку утвореного метилтрифенілового Детритилювання в метанолі, саме без додаетеру формули XIII і потім дебензилюють до валвання будь-якого каталізатора, відбувається шлязартану формули VIII, використовуючи один з опихом перемішування відповідної тритильованої саних способів. проміжної сполуки з метанлом при температурі між 20°С та температурою кипіння метанолу, пе 9 86947 10 Якщо вихідною проміжною сполукою є тритилоб'єму і осаджений після охолодження метилтрикандезартан формули XI, фільтрат, одержаний фенілметиловий етер (XIII) відсмоктували та пропісля відсмоктування метилтрифенілметилового мивали невеликою кількістю охолодженого льодом етеру, випаюють до сухого залишку і потому отриметанолу. Отримували 3,71г (90%) метилтрифенімують кандезартан цилексетил формули XII крислметилового етеру (XIII). Фільтрат випарювали і талізацією з відповідного розчинника. Альтернатизалишок після випарювання розчиняли в 100мл вно, метилтрифеніловий етер може бути метанолу. Додавали 1,50г КНСО3 і суміш кип'ятили видалений кристалізацією продукту з придатного зі зворотним холодильником протягом 4 годин. розчинника, переважно, з циклогексану, або з суПотім метанол випарювали і після додавання ацеміші придатних розчинників. Суміші розчинників, в тону залишок після випарювання кристалізували. яких кандезартан цилексетил легко розчиняється, Кристалі відсмоктували і промивали невеликою з розчинниками, в яких ця речовина розчиняється кількістю охолодженого льодом ацетону. Отримутільки частково, вважаються найкращими змішавали 5,29г (76,5%) калієвої солі 2-бутил-4-хлор-1ними розчинниками. Розчинниками, в яких канде[(2'-трифенілметилтетразол-5-іл)-1,1′-біфеніл-4-іл]зартан цилексетил легко розчиняється і які можуть 5-(гідроксиметил)імідазолу (III) Т.пл. (ДСК - дифевикористовуватися, є С1-С4 спирти, переважно ренційна сканувальна калориметрія) 229,7°С (зміметанол, етанол або 2-пропанол, С1-С2 галогенона кристалічної форми) і 274,6°С. 1Н ЯМР спектри вані розчинники, переважно дихлорметан і хлоро(ДМСО): 0,83 т, J=7,27, 3Н; 1,26 м, 2Н; 1,48 м, 2Н; форм, С1-С4 аліфатичні кетони, переважно ацетон 2,51 т; J=7,53 , 2Н; 4,34 с, 2Н; 5,23 с, 2Н; 6,93 д, або 2-бутанон, діалкілові етери з С1-С4 алкілами, 7=8,36, 2Н; 7,13 d, J=8,34, 2Н; 7,32-7,39 м, 3Н; 7,55 переважно діізопропіловий етер і метил третм, 1Н. бутиловий етер, та естери С1-С5 карбонових кисПриклад 3 лот з С1-С4 аліфатичними спиртами, переважно Калієва сіль 2-бутил-4-хлор-1 -[(2'-тетразол-5метилацетат, етилацетат, ізопропілацетат або іл )-біфеніл-4-іл]-5-(гідроксиметил)імідазолу (лозаетилпропіонат. Розчинниками, в яких кандезартан ртан, II) цилексетил розчиняється лише частково і які мо2,10г Кальцинованого карбонату калію жуть використовуватися, є циклоалкани, напри(0,0150моль) пододавали до суспензії 10г клад, циклогексан, С5-С8 аліфатичні вуглеводні, (0,0150моль) 2-бутил-4-хлор-1-[(2′-1-тритил-1Ннаприклад, пентан, гексан, гептан або ізооктан. тетразол-5-іл)-1,1'-біфеніл-4-іл]-5Винахід пояснюється більш детально наступ(гідроксиметил)імідазол (тритиллозартан, IV) в ними робочими прикладами. Ці приклади, які ілюс65мл безводного метанолу, і суміш піддавали китрують удосконалення способу згідно з винахоп'ятінню зі зворотним холодильником. Суміш після дом, є лише ілюстративними за призначенням і 6 годин кип'ятіння перемішували протягом ночі ніяким чином не обмежують об'єм винаходу. нагрівання. Наступного дня розчин концентрували Приклади до 1/3 його об'єму і після осадження при охолоПриклад 1 дженні метилтрифенілметиловий етер (XIII) від2-Бутил-4-хлор-1-[[(2'-тетразол-5-іл)-1,1'смоктували. Фільтрат випарювали, і залишок після біфеніл-4-іл]метил]-5-гідроксиметил імідазол випарювання кристалізувався після додавання Суспензію 10г (0,015моль) 2-бутил-4-хлор-1ацетону. Кристали відсмоктували і промивали не[[(2'-1-тритил-1Н-тетразол-5-іл)-1,1-біфеніл-4великою кількістю охолодженого льодом ацетон. іл]метил]-5-гідроксиметилімідазолу (тритиллозарБуло отримано 4,98г (72.0%) калієвої солі 2-бутилтану, IV) в 50мл безводного метанолу кип'ятили зі 4-хлор-1-[(2'-трифенілметилтетразол-5-іл)-1,1'зворотнім холодильником протягом 7 годин. Розбіфеніл-4-іл]-5-(гідроксиметил)імідазолу (III). Т.пл. чин потім охолоджували до -10°С і перемішували (ДСК) 233,9°С (зміна кристалічної форми) і при цій температурі протягом ночі, осаджені крис273,5°С. тали відсмоктували і промивали невеликою кількіПриклад 4 стю охолодженого льодом метанолу. Отримували Калієва сіль 2-бутил-4-хлор-1-[(2'-тетразол-53,7г (90%) метилтрифенілметилового етеру (XIII). іл)-1,1'-біфеніл-4-іл]-5-(гідроксиметил)імідазол (лоОб'єднані маткові розчини випарювали і кип'ятили зартан, III) з 50мл гексану, суміш охолоджували і нерозчинну 2,10г Кальцинованого карбонату калію частину відсмоктували, перемішували при кімнат(0,0150моль) додавали до суспензії 10г ній температурі з 50мл циклогексану протягом 10 (0,0150моль) 2-бутил-4-хлор-1-[(24-тритил-1Нгодин, нерозчинну частину відсмоктували. Отритетразол-5-іл)-біфеніл-4-іл]-5мували 6,2г продукту (98%) з т.пл. 186-188°С. 1Н (гідроксиметил)імідазолу (тритиллозартан, IV) в ЯМР спектри (ДМСО): 0,81 τ, J= 7,24, 3Н; 1,27 м, 65мл безводного метанолу і суміш піддавали ки2Н; 1,47 м, 2Н; 2,47 т, J= 7,57,2Н; 4,35 с, 2Н; 5,26 с, п'ятінню зі зворотним холодильником. Суміш після 2Н; 7,03-7,12 м, 4Н; 7,49-7,73 м, 4Н. 5 годин кип'ятіння перемішували протягом ночі. Приклад 2 Наступного дня розчин концентрували до 1/3 його Калієва сіль 2-бутил-4-хлор-1-[(2'-тетразол-5об'єму і охолоджували, осаджений метилтрифенііл)-1,1'-біфеніл-4-іл]-5-(гідроксиметил)імідазолу лметиловий етер (XIII) відсмоктували. Фільтрат (лозартан, III) випарювали, залишок після випарювання розчиСуспензію 10г (0.015моль) 2-бутил-4-хлор-1няли в 30мл ізопропілового спирту і до одержаного [(2'-1-тритил-1Н-тетразол-5-іл)-1,1-біфеніл-4-іл]-5розчину додавали 70мл циклогексану. Осаджені (гідроксиметил)імідазолу (тритиллозартану, IV) в кристали відсмоктували і промивали невеликою 100мл безводного метанолу кип'ятили протягом 7 кількістю охолодженого льодом ацетону. Одержугодин. Розчин потім концентрували до 1/5 його вали 5,50г (79,5%) калієвої солі 2-бутил-4-хлор-1 11 86947 12 [(2'-трифенілметилтетразол-5-іл)-1,1'-біфеніл-4-іл]додавання ацетону. Кристали відсмоктували і 5-(гідроксиметил)імідазолу (III). Т.пл. (ДСК) промивали невеликою кількістю охолодженого 232,7°С (зміна кристалічної форми) і 272,9°С. льодом ацетону. Отримували 6,31г (91,2%) калієПриклад 5 вої солі 2-бутил-4-хлор-1-[(2'-1Н-тетразол-5Калієва сіль бутил-4-хлор-1-[(2'-1Н-тетразол-5іл)біфеніл-4-іл]-5-(гідроксиметил)імідазолу (ІІІ). іл)біфеніл-4-іл]-5-(гідроксиметил)імідазолу (лозарТ.пл. (ДСК) 229,9°С (зміна кристалічної форми) і тан, III) 274,2°С. 1,05г Кальцинованого карбонату калію Приклад 8 (0,0075моль) додавали до суспензії 10г Калієва сіль 2-бутил-4-хлор-1-[(2'-1Н-тетразол(0,015моль) 2-бутил-4-хлор-1-[(2'-1 -тритил-1Н5-іл)біфеніл-4-іл]-5-(гідроксиметил)імідазолу (лотетразол-5-іл)біфеніл-4-іл]-5зартан, III) (гідроксиметил)імідазолу (тритиллозартан, IV) в 1,52г Гідрокарбонату калію (0,0150моль) до65мл безводного етанолу і суміш піддавали кип'ядавали до суспензії 10г (0,015моль) 2-бутил-4тінню зі зворотним холодильником на масляній хлор-1-[(2'-1-тритил-1Н-тетразол-5-іл)-1,1'-біфенілбані. Нагрівання припиняли через 8 годин, і суміш 4-іл]-5-(гідроксиметил)-імідазолу (тритиллозартан, перемішували протягом ночі. Наступного дня розIV) в 65мл безводного метанолу і суміш піддавали чин концентрували до 1/3 його об'єму і охолоджукип'ятінню зі зворотним холодильником. Нагріванвали, осаджений метилтрифенілметиловий етер ня припиняли через 6 годин кип'ятіння, і суміш (XIII) відсмоктували. Фільтрат випарювали, і залиперемішували протягом ночі. Наступного дня розшок після випарювання кристалізувався після дочин концентрували до 1/3 його об'єму і осаджений давання ацетону. Кристали відсмоктували і пропісля охолодження метилтрифенілметиловий етер мивали невеликою кількістю охолодженого льодом (XIII) відсмоктували. Фільтрат випарювали, і після ацетону. Було одержано 4,98г (72,0%) калієвої солі додавання ацетону залишок кристалізувався. Кри2-бутил-4-хлор-1-[(2'-1Н-тетразол-5-іл)біфеніл-4стали відсмоктували і промивали невеликою кільіл]-5-(гідроксиметил)імідазолу (III). Т.пл. (ДСК) кістю охолодженого льодом ацетону. Отримували 234,1°С (зміна кристалічної форми) і 275,2°С. 6,36г (91,9%) Приклад 6 калієвої солі 2-бутил-4-хлор-1-[(2'-1НКалієва сіль 2-бутил-4-хлор-1-[(2'-1Н-тетразолтетразол-5-іл)біфеніл-4-іл]-5-(гідроксиметил)5-іл)біфеніл-4-іл]-5-(гідроксиметил)імідазолу (лоімідазолу (III). Т.пл. (ДСК) 232,9°С (зміна кристалізартан, III) чної форми) і 274,5°С. 1,05г Кальцинованого карбонату калію Приклад 9 (0,0075моль) додавали до суспензії 10г 2-Бутил-3-[[2'-(1 Н-тетразол-5-іл)[1,1'-біфеніл](0,015моль) 2-бутил-4-хлор-1-[(2'-1-тритил-1Н4-іл]метил-1,3-діазаспіро[4.4]нон-1-єн (ірбезартан, тетразол-5-іл)-1,1'-біфеніл-4-іл]-5VI) (гідроксиметил)імідазолу (тритиллозартан, IV) в Суспензію 1г (0,0015моль) 2-бутил-3-[2′-(165мл безводного метанолу і суміш піддавали китритил-1Н-тетразол-5-іл)-1,1′-біфеніл-4-ілметил]п'ятінню зі зворотним холодильником на масляній 1,3-діазаспіро[4.4]нон-1-ен-4-ону (тритилірбезарбані. Нагрівання припиняли через 8 годин, і суміш тан, VII) в 10мл безводного метанолу кип'ятили зі перемішували протягом ночі. Наступного дня роззворотним холодильником протягом 10 годин. Почин концентрували до 1/3 його об'єму і охолоджутім розчин охолоджували до -10°С і перемішували вали, осаджений метилтрифенілметиловий етер при цій температурі протягом ночі; осаджені крис(XIII) відсмоктували. Фільтрат випарювали, залитали відсмоктували і промивали невеликою кількішок після випарювання розчиняли в 30мл ізопростю охолодженого льодом метанолу. Отримували пілового спирту і додавали 70мл циклогексану. 0,30г (73%) метилтрифенілметилового етеру (XIII). Осаджені кристали відсмоктували і промивали Об'єднані маткові розчини випарювали. Одержаневеликою кількістю охолодженого льодом ацетоний сирий ірбезартан (VI) кристалізували з ізопрону. Було отримано 6,12г (88,5%) калієвої солі 2панолу і промивали гексаном. Було отримано 0,45г бутил-4-хлор-1-[(2'-1Н-тетразол-5-іл)біфеніл-4-іл](71%) ірбезартану (VI). Т.пл. = 180 °С-181°С. 5-(гідроксиметил)імідазолу (III). Т.пл. (ДСК) Приклад 10 229,1°С (зміна кристалічної форми) і 271,8°С. N-(1-Оксопентил)-N-[[2'-(1Н-тетразол-5-іл)[1,1'Приклад 7 біфеніл]-4-іл]метил]-L-валін (валзартан, VIII) Калієва сіль 2-бутил-4-хлор-1-[(2'-1Н-тетразолСуспензію 10г (0,013моль) бензилового естеру 5-іл)біфеніл-4-іл]-5-(гідроксиметил)імідазолу (лоN-(1-оксопентил)-N-[[2'-(1-тритил-1Н-тетразол-5зартан, III) іл)[1,1'-біфеніл]-4-іл]метил]-L-валіну (бензилового 1,52г Гідрокарбонату калію (0,0150моль) поестеру тритилвалзартану, IX) в 75мл безводного додавали до суспензії 10г (0,015моль) 2-бутил-4метанолу кип'ятили зі зворотним холодильником хлор-1-[(2-1-тритил-1Н-тетразол-5-іл)-1,1'-біфенілпротягом 10 годин. Потім розчин охолоджували до 4-іл]-5-(гідроксиметил)імідазолу (тритиллозартан, -10°С і перемішували при цій температурі протяIV) в 65мл безводного метанолу, і суміш піддавали гом ночі; осаджені кристали відсмоктували і прокип'ятінню зі зворотним холодильником. Нагріванмивали невеликою кількістю охолодженого льодом ня припиняли через 6 годин кип'ятіння, і суміш метанолу. Отримували 3г (84%) метилтрифенілперемішували протягом ночі. Τ Наступного дня метилового етеру (XIII). Потім сирий бензиловий розчин концентрували до 1/3 його об'єму і осаджеестер вапзартану (X) розчиняли в 20мл метанолу і ний після охолодження метилтрифенілметиловий гідрували на 3% Pd/C. Матковий розчин після виетер (XIII) відсмоктували. Фільтрат випарювали, і далення каталізатора випарювали до сухого зазалишок після випарювання кристалізувався після лишку і після кристалізації з суміші етилаце 13 86947 14 тат/циклогексан отримували 3г (53%) кристалізоЗалишок після випарювання (1,5г), отриманий ваного валзартану (VIII). Т.пл. = 109°С-113°С. способом, описаним в прикладі 12, розчиняли в Приклад 11 невеликій кількості 2-пропанолу, і після додавання гексану осаджувалось 1,25г (87%) білого порошкоN-(1-Оксопентил)-N-[[2′-(1Н-тетразол-5-іл)[1,1'подібного продукту. біфеніл]-4-іл]метил]-L-валін (валзартан, VIII) Приклад 14 Суспензію 10г (0,013моль) бензилового естеру 1-(Циклогексилоксикарбонілокси)етил-2N-(1-оксопентил)-N-[[2'-(1-тритил-1H-тетразол-5етокси-1-[[2'-(1Н-тетразол-5-іл)біфеніл-4іл)[1,1'-біфеніл]-4-іл]метил]-L-валіну (бензилового іл]метил]бензімідазол-7-карбоксилат (XII) естеру тритилвалзартану, IX) в 75мл безводного Залишок після випарювання (1,5г), отриманий метанолу кип'ятили зі зворотним холодильником способом, описаним в прикладі 12, розчиняли в протягом 10 годин. Потім розчин охолоджували до невеликій кількості дихлорметану, і після додаван-10°С і перемішували при цій температурі протяня гексану осаджувалось 1,3г (91%) білого порошгом ночі; осаджені кристали відсмоктували і прокоподібного продукту. мивали невеликою кількістю охолодженого льодом Приклад 15 метанолу. Отриманий таким чином метанольний 1-(Циклогексилоксикарбонілокси)етил-2розчин, після додавання гідроксиду калію (0,6г), етокси-1-[[2'-(1Н-тетразол-5-іл)біфеніл-4кип'ятили зі зворотним холодильником протягом 4 іл]метил]бензімідазол-7-карбоксилат (ХІІ) годин. Метанол випарювали у вакуумі, суміш розЗалишок після випарювання (1,5г), отриманий бавляли 10мл води і, після підкислення соляною способом, описаним в прикладі 12, розчиняли в кислотою, валзартан екстрагували з використанневеликій кількості ацетону, і після додавання гекням етилацетату (3 х 40мл). Органічний шар просану осаджувалось 1,28г (90%) білого порошкопомивали водою (2 х 25мл), концентрували до 30мл і дібного продукту. після додавання циклогексану (50мл) продукт криПриклад 16 сталізували. Після відсмоктування та сушіння у 1-(Циклогексилоксикарбонілокси)етил-2вакуумі отримували 3,5г (62%) валзартану (VIII). етокси-1-[[2'-(1Н-тетразол-5-іл)біфеніл-4Т.пл. = 109-113°С. іл]метил]бензімідазол-7-карбоксилат (ХІІ) Приклад 12 Залишок після випарювання (1,5г), отриманий 1-(Циклогексилоксикарбонілокси)етил-2способом, описаним в прикладі 12, розчиняли в етокси-1-[[2'-(1Н-тетразол-5-іл)біфеніл-4невеликій кількості метил трет-бутилового етеру і іл]метил]бензімідазол-7-карбоксилат(ХІІ) потому додавали гептан для того, щоб досягти Суміш 1-(циклогексилоксикарбонілокси)етил-2мутності. Після нагрівання утворювався прозорий етокси-1-[[2'-(N-трифенілметил-1Н-тетразол-5розчин, з якого, після охолодження та затравки іл)біфеніл-4-іл]метил]бензімідазол-7-карбоксилату кристалами, отриманими способом за прикладом (XI) (2г) та метанолу (40мл) перемішували кип'яти12, отримували 1,2г (84%) білого кристалічного ли зі зворотним холодильником протягом 24 годин. продукту. Одержаний розчин концентрували до 1/4 його об'Приклад 17 єму і осаджені після охолодження кристали від1-(Циклогексилоксикарбонілокси)етил-2смоктували і промивали невеликою кількістю (0,5г) етокси-1-[[2'-(1Н-тетразол-5-іл)біфеніл-4метанолу, охолодженого до 0°С. Матковий розчин іл]метил]бензімідазол-7-карбоксилат (XIІ) випарювали (1,5г) і після кристалізації з циклогекЗалишок після випарювання (1,5г), отриманий сану отримували 1,1г (76%) продукту у формі біспособом, описаним в прикладі 12, розчиняли в лих кристалів. 1 невеликій кількості 2-бутанону і потім додавали Н ЯМР (250 МГц, CDCI3) δ: 1,13-1,50 (12Н, м); ізооктан для того, щоб досягти мутності. Після 1,64 (2Н, м); 1,79 (2Н, м); 4,10450 (3Н, м); 5,62 (2Н, нагрівання утворювався прозорий розчин, з якого, д); 6,65-6,93 (7Н, м); 7,27-7,28 (1Н, м); 7,46-7,48 після охолодження та затравки кристалами, отри(1Н, м); 7,56-7,59 (2Н, м); 7,98-8,02 (1Н,м). маними способом за прикладом 12, отримували Приклад 13 1,2г (84%) білого кристалічного продукту. 1-(Циклогексилоксикарбонілокси)етил-2етокси-1-[[2'-(1Н-тетразол-5-іл)біфеніл-4іл]метил]бензімідазол-7-карбоксилат (ХІІ) Комп’ютерна верстка Л. Купенко Підписне Тираж 28 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for the production of losartan
Автори англійськоюRadl Stanislav, Stach Jan, Klecan Ondrej
Назва патенту російськоюСпособ получения лозартрана
Автори російськоюРадл Станислав, Стах Ян, Клецан Ондрей
МПК / Мітки
МПК: C07D 257/00, C07D 403/10
Мітки: спосіб, одержання, лозартрану
Код посилання
<a href="https://ua.patents.su/7-86947-sposib-oderzhannya-lozartranu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання лозартрану</a>
Спосіб одержання лозартану калію

Номер патенту: 72803
Опубліковано: 15.04.2005
Автори: Немеш Андраш, Фішер Янош, Цібула Ласло, Надіне Багді Юдіт, Хегедюш Іштван, Фаркаш Еньоне, Дешне Юхас Іда, Балло Ілдіко, Петені Ендрене, Креідл Янош, Веркне Папп Ева
МПК: C07D 403/10, A61P 9/12
Мітки: калію, одержання, спосіб, лозартану
Формула / Реферат:
1. Спосіб одержання лозартану калію формули (І) (І)хімічна назва якого 2-н-бутил-4-хлор-1-[(2'-(тетразол-5-іл)-1,1'-біфеніл-4-іл)метил]імідазол-5-метанол калію, із використанням як вихідної сполуки 2-н-бутил-4-хлор-1-[(2'-(2-трифенілметил-2Н-тетразол-5-іл)-1,1'-біфеніл-4-іл)метил]-1Н-імідазол-4-метанолу формули (III)
Заміщені фенільні похідні, спосіб їх одержання (варіанти), фармацевтична композиція на їх основі (варіанти)

Номер патенту: 73284
Опубліковано: 15.07.2005
Автори: Даль Б'ярне Г., Хрістоферсен Палле
МПК: C07C 275/42, A61P 35/04, A61K 31/4439, A61P 19/08, A61P 1/12, A61P 1/04, A61K 31/41, C07D 233/34, C07D 233/32, A61P 27/06, A61P 17/02, C07C 275/30, C07D 257/00, A61P 29/00, C07D 401/12, A61P 43/00, A61P 9/10, A61P 19/10, A61K 31/496, C07C 273/18, A61P 7/06, A61K 31/17, A61P 37/08, A61K 31/4166, A61K 31/192, A61P 35/00, A61P 9/12
Мітки: варіанти, спосіб, похідні, фенільні, основі, фармацевтична, композиція, заміщені, одержання
Формула / Реферат:
1. Сполука формулиабо її фармацевтично прийнятна сіль, де R1 - тетразоліл,R4, R5 та R2 кожний незалежно вибрано з гідрогену, алкілу, алкокси, гідрокси, галогену, трифлуорметилу, ціано, нітро, аміно, алкіламіно, NHCOR9, CO2R9, -CON(R9)2, -NHSO2-R9, -SO2N(R9)2 або арилу, як варіант, заміщеного CON(R9)2, NHCOR9, SO2N(R9)2, CO2R9, де R9 - гідроген,...
Акрилоїлзаміщенa похідна дистаміцину, спосіб її одержання (варіанти) та фармацевтична композиція на її основі

Номер патенту: 61922
Опубліковано: 15.12.2003
Автори: Францетті Крістіна, Кодзі Паоло, Беріа Італо, Каполонго Лаура, Калдареллі Маріна, Бьясолі Джованні
МПК: A61P 35/00, C07D 207/34, A61K 31/40, A61P 31/12
Мітки: фармацевтична, композиція, варіанти, похідна, основі, одержання, дистаміцину, акрилоїлзаміщенa, спосіб
Формула / Реферат:
1. Акрилоїлзаміщена похідна дистаміцину формули:,де:n означає 2, 3 або 4;R1 або R2 вибирають, кожен незалежно, з: водню, галогену і С1-С4 алкілу;R3 означає водень або галоген;В вибирають з:де R4, R5, R6, R7 і R8 означають, кожен незалежно, водень або С1-С4 алкіл, за умови, що, принаймні, один з R4, R5 і R6 означає С1-С4 алкіл, або її фармацевтичнo прийнятна сіль.2. Сполука за п....
Похідні цинаміду, корисні при лікуванні запальних та імунних захворювань, композиція, що містить такі похідні, і спосіб одержання похідних цинаміду

Номер патенту: 72909
Опубліковано: 16.05.2005
Автори: Гунавардана Індрані В., Бойд Стівен А., Ліу Ганг, Лінч Джон К., Джае Хван-Су, Жу Гуі-Донг, фон Гельдерн Том, Ксін Жілі, Стіджер Майкл А., Пей Жонгуа, Фрімен Дженіфер С., Лінк Джеймс, Уінн Мартін
МПК: C07D 209/12, C07D 209/08, C07D 211/60, C07D 207/263, C07D 211/46, A61K 31/417, C07D 241/04, C07D 295/185, C07D 207/09, C07D 295/215, C07D 241/18, A61K 31/506, C07D 241/24, C07D 213/81, C07D 405/12, C07D 215/36, C07D 213/82, C07D 211/62, C07D 233/61, C07D 413/04, C07D 207/08, C07D 413/14, C07D 417/12, C07D 239/42, C07D 401/06, A61K 31/404, C07D 209/18, A61K 31/438, C07D 207/273, A61K 31/5375, C07D 295/16, A61K 31/165, C07D 207/22, C07D 207/36, C07D 207/27, C07D 207/20, C07C 323/63, A61K 31/407, A61K 31/454, A61K 31/495, C07D 265/30, C07D 211/44, C07D 307/68, C07D 491/10, C07D 413/12, C07D 213/74, C07D 211/74, C07D 207/14, A61K 31/443, C07D 211/22, A61P 37/02, A61K 31/44, C07D 217/06, A61K 31/40, A61K 31/167, C07D 207/26, C07D 215/04, C07D 211/42, C07D 401/12, C07D 295/26, C07D 307/52, C07D 487/08, A61K 31/445, A61K 31/472, A61K 31/5377, C07C 323/62, A61P 29/00, C07D 317/62, C07D 207/34, C07D 235/26, A61K 31/16, C07D 401/04, A61K 31/496, C07D 243/08, C07D 209/14, C07D 211/26, C07D 317/58, C07D 319/00, C07D 207/12, C07D 211/66, C07D 411/00, A61K 31/54, C07D 405/14, A61K 31/45, A61K 31/541, A61K 31/164, A61K 31/4468, A61K 31/4465, C07D 295/12, C07D 403/04, C07D 295/205, C07D 295/13, C07D 211/54, C07D 213/75, C07D 295/32, C07D 295/18, C07D 405/06, C07D 403/12, A61K 31/453, A61K 31/4015, C07D 471/10
Мітки: лікуванні, одержання, похідні, захворювань, імунних, запальних, цинаміду, корисні, спосіб, такі, похідних, композиція, містить
Формула / Реферат:
1. Похідні цинаміду формули I:або їх фармацевтично прийнятна сіль або пролікарська форма, в якій R1, R2, R3, R4 і R5 незалежно вибирають з a) водню,b) галогену,c) алкілу,d) галогеналкілу,e) алкокси,f) ціано,g) нітро,h) карбоксальдегіду, іпри умові, що принаймні один R1 або R3 є...
Спосіб одержання похідних хіноліну

Номер патенту: 78310
Опубліковано: 15.03.2007
Автор: Янссон Карл
МПК: C07D 215/56, A61K 31/47, A61P 37/00, A61P 35/00, C07D 491/04
Мітки: похідних, одержання, спосіб, хіноліну
Формула / Реферат:
1. Спосіб одержання сполуки загальної формули (І), (І)деR вибирається з групи: метил, етил, н-пропіл, ізо-пропіл, н-бутил, ізо-бутил, втор(sec)-бутил та аліл;R5 вибирається з групи: метил, етил, н-пропіл, ізо-пропіл, метокси, етокси, метилтіо, етилтіо, н-пропілтіо, метилсульфініл, етилсульфініл, фтор, хлор, бром, трифторметил та...